Introduction
-
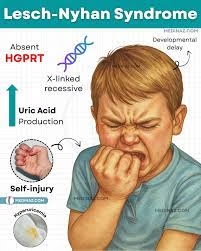
Lesch–Nyhan Syndrome (LNS) is a rare inherited metabolic disorder that primarily affects males and is caused by a defect in purine metabolism.
-
It results from a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT or HGPRT), which is essential for the salvage pathway of purine nucleotide synthesis.
-
Due to an enzyme deficiency, uric acid accumulates in the body, leading to hyperuricemia, gout-like symptoms, and the formation of kidney stones.
-
The condition is inherited as an X-linked recessive trait, meaning the defective gene (HPRT1) is located on the X chromosome (Xq26–Xq27 region).
-
Only males are usually affected because they have a single X chromosome, while females act as carriers and rarely show symptoms.
-
It was first described in 1964 by Michael Lesch and William Nyhan, who noticed a combination of neurological abnormalities, overproduction of uric acid, and self-mutilating behavior in affected children.
-
The syndrome presents with a triad of features:
-
Overproduction of uric acid (leading to gout and renal stones)
-
Neurological dysfunction (spasticity, dystonia, choreoathetosis)
-
Behavioral disturbances (especially self-injurious behavior)
-
-
Neurological symptoms occur due to dopamine deficiency in the basal ganglia, linking biochemical enzyme defects with neurobehavioral outcomes.
-
The disease is often recognized in infancy or early childhood when developmental delay and uric acid crystals in urine (orange sand) become apparent.
-
There is no curative treatment, but early diagnosis, symptomatic management (e.g., allopurinol), physiotherapy, and genetic counseling help improve the quality of life and prevent complications.
-
Lesch–Nyhan Syndrome is a classic example of how a single gene defect can result in multisystemic biochemical, neurological, and behavioral abnormalities.
Biochemical Basis and Function of HPRT
Under normal conditions:
-
Purines (adenine, guanine, hypoxanthine) are salvaged via the HPRT enzyme.
-
HPRT catalyzes:
-
Hypoxanthine + PRPP → IMP + PPi
-
Guanine + PRPP → GMP + PPi
(PRPP = Phosphoribosyl pyrophosphate)
-
In Lesch–Nyhan Syndrome:
-
The lack of HPRT blocks the salvage pathway, causing an accumulation of hypoxanthine and guanine.
-
These are degraded by xanthine oxidase into uric acid, leading to hyperuricemia.
-
Elevated PRPP levels trigger de novo purine synthesis, further amplifying uric acid production — a biochemical vicious cycle.
Genetics and Molecular Pathogenesis
-
Gene name: HPRT1
-
Chromosomal location: Xq26–Xq27
-
Type of inheritance: X-linked recessive
-
Mutation types: Point mutations, deletions, insertions, or duplications leading to complete or partial enzyme deficiency.
Males (XY) with the defective HPRT1 gene develop the full syndrome, while female carriers (XX) are usually asymptomatic due to lyonization (random X-inactivation) but may show mild symptoms in rare cases.
At the molecular level, absence of HPRT affects purine recycling in neurons, resulting in:
-
Reduced dopamine synthesis in the basal ganglia (especially striatum).
-
Functional impairment in dopaminergic neurotransmission causes motor dysfunction, dystonia, and compulsive self-mutilation.
Pathophysiology
The multisystem effects arise from two primary mechanisms:
-
Metabolic Disturbance:
-
Accumulation of uric acid → Gout, nephrolithiasis, hematuria, renal failure.
-
Depletion of guanine/hypoxanthine bases → altered nucleotide balance.
-
-
Neurochemical Disturbance:
-
Decreased dopamine in the basal ganglia.
-
Abnormal brain development and synaptic signaling.
-
Leads to hyperkinesia, spasticity, dystonia, and behavioral outbursts.
-
Clinical Manifestations
| System Involved | Clinical Features |
|---|---|
| Neurological | Developmental delay, hypotonia → later spasticity, dystonia, choreoathetosis, opisthotonus, dysarthria, pyramidal signs |
| Behavioral | Self-injurious behavior (lip/finger biting), aggression, compulsive actions, agitation, frustration intolerance |
| Metabolic / Renal | Hyperuricemia, orange sand-like crystals in urine, gouty arthritis, nephrolithiasis, renal colic |
| Musculoskeletal | Contractures, deformities due to chronic spasticity |
| Growth and Cognition | Poor growth, intellectual disability (mild–moderate), failure to thrive |
| Other Features | Anemia, megaloblastic changes, facial injuries due to self-biting |
Diagnosis
1. Clinical Clues
-
Male infant with developmental delay and orange crystalline deposits in urine.
-
Onset of self-biting behaviour by 2–3 years.
-
Neurological hypertonia and movement abnormalities.
2. Laboratory Investigations
| Test | Findings |
|---|---|
| Serum uric acid | Markedly elevated (>7 mg/dL) |
| Urine uric acid/creatinine ratio | Increased |
| Enzyme assay (HPRT activity) | Absent or <1% of normal in erythrocytes or fibroblasts |
| PRPP concentration | Elevated |
| Lactate/Ammonia | Usually normal (helps rule out other metabolic disorders) |
3. Genetic Diagnosis
-
Molecular analysis of HPRT1 gene via PCR and sequencing.
-
Carrier detection in females through heterozygosity testing.
-
Prenatal diagnosis by:
-
Chorionic villus sampling (10–12 weeks)
-
Amniocentesis (16–18 weeks)
-
Enzyme activity or molecular mutation detection.
-
4. Imaging Findings
-
Brain MRI/CT: Atrophy of the basal ganglia (caudate nucleus and putamen).
-
Renal ultrasound: Detects uric acid stones or nephrocalcinosis.
Management and Treatment
There is currently no curative therapy; however, symptom-oriented, multidisciplinary management is essential.
1. Pharmacologic Management
| Drug | Purpose |
|---|---|
| Allopurinol | Inhibits xanthine oxidase → lowers uric acid levels, prevents gout and renal stones. |
| Baclofen / Diazepam / Clonazepam | Reduces spasticity and dystonia. |
| Gabapentin / Antipsychotics (Risperidone, Haloperidol) | Controls aggression, self-mutilation, and compulsive behaviors. |
| Levodopa / Dopamine agonists | Under study to correct dopamine deficiency (variable results). |
⚠️ Allopurinol does not improve neurological symptoms—it only prevents metabolic complications.
2. Physical and Supportive Therapy
-
Physiotherapy to prevent contractures.
-
Occupational therapy for functional mobility.
-
Speech therapy for dysarthria.
-
Dental and protective care: Mouth guards, gloves, and restraints to prevent self-injury.
3. Nutritional and Hydration Support
-
Low-purine diet (avoid red meat, organ meat, fish).
-
Adequate water intake to prevent urate crystallisation.
-
Monitor kidney function periodically.
4. Genetic Counselling
-
Essential for families with a previous case.
-
Counselling for female carriers to discuss recurrence risk (50% chance for affected sons).
Prognosis
-
Without treatment, patients may develop renal failure and gouty arthritis by adolescence.
-
With modern supportive care and uric acid control, survival may extend into the 3rd or 4th decade.
-
Neurological disability and self-mutilation persist lifelong.
-
Quality of life improves significantly with early diagnosis, physical therapy, and behavioral support.
Recent Research Directions
-
Gene Therapy: Attempts to introduce functional HPRT1 gene using viral vectors.
-
Stem Cell Therapy: Exploring replacement of dopaminergic neurons in basal ganglia.
-
CRISPR-based correction: Targeted editing of HPRT1 gene in vitro.
-
Enzyme Replacement and Chaperone Therapy: Experimental approaches to stabilize residual HPRT enzyme.
-
Neurotransmitter Modulation: Trials on L-DOPA, 5-HTP, and dopamine receptor agonists.
MCQs
-
Lesch–Nyhan Syndrome is caused by a deficiency of which enzyme?
A. Xanthine oxidase
B. Adenine phosphoribosyltransferase
C. Hypoxanthine-guanine phosphoribosyltransferase
D. Glucose-6-phosphatase -
The inheritance pattern of Lesch–Nyhan Syndrome is:
A. Autosomal dominant
B. Autosomal recessive
C. X-linked recessive
D. Y-linked -
Lesch–Nyhan Syndrome primarily affects which gender?
A. Only females
B. Only males
C. Both equally
D. Predominantly females -
The defective gene in Lesch–Nyhan Syndrome is located on which chromosome?
A. Chromosome 7
B. Chromosome 12
C. Chromosome X (Xq26–Xq27)
D. Chromosome Y -
The primary biochemical abnormality in Lesch–Nyhan Syndrome is:
A. Hyperglycemia
B. Hyperlipidemia
C. Hyperuricemia
D. Hypocalcemia -
Which of the following pathways is affected in Lesch–Nyhan Syndrome?
A. Glycolysis
B. Purine salvage pathway
C. Urea cycle
D. Citric acid cycle -
Which enzyme converts hypoxanthine to IMP in the salvage pathway?
A. Xanthine oxidase
B. HPRT
C. Adenylate kinase
D. Guanine deaminase -
What type of neurological disturbance is most characteristic of Lesch–Nyhan Syndrome?
A. Parkinsonism
B. Dystonia and spasticity
C. Epilepsy
D. Ataxia -
The self-mutilation behavior in Lesch–Nyhan Syndrome is due to deficiency of which neurotransmitter?
A. Serotonin
B. Dopamine
C. Acetylcholine
D. GABA -
The orange-colored crystals seen in urine of affected infants are composed of:
A. Calcium oxalate
B. Sodium urate
C. Cystine
D. Urea -
Which of the following is NOT a feature of Lesch–Nyhan Syndrome?
A. Self-mutilation
B. Mental retardation
C. Hypouricemia
D. Gout -
The classic triad of Lesch–Nyhan Syndrome includes:
A. Anemia, jaundice, hepatomegaly
B. Hyperuricemia, neurological dysfunction, self-mutilation
C. Hyperglycemia, ketosis, dehydration
D. Hypocalcemia, tetany, muscle weakness -
Which scientist(s) first described Lesch–Nyhan Syndrome?
A. Seegmiller and Wyngaarden
B. Lesch and Nyhan
C. Watson and Crick
D. Sanger and Kendrew -
Allopurinol is used in Lesch–Nyhan Syndrome to:
A. Increase uric acid excretion
B. Inhibit uric acid synthesis
C. Increase dopamine synthesis
D. Replace enzyme activity -
Allopurinol acts by inhibiting which enzyme?
A. Xanthine oxidase
B. HPRT
C. Adenosine deaminase
D. PRPP synthetase -
Which of the following complications can result from hyperuricemia in Lesch–Nyhan Syndrome?
A. Gouty arthritis
B. Kidney stones
C. Both A and B
D. None of the above -
Which organ system is primarily responsible for neurological symptoms in Lesch–Nyhan Syndrome?
A. Cerebellum
B. Basal ganglia
C. Brainstem
D. Cerebral cortex -
The abnormal behavior of self-biting in Lesch–Nyhan Syndrome is termed:
A. Autophagia
B. Cannibalism
C. Coprophagia
D. Pica -
The immediate biochemical cause of increased uric acid formation is:
A. Decreased breakdown of purines
B. Increased de novo purine synthesis
C. Decreased pyrimidine synthesis
D. Defective urea cycle -
Which laboratory test helps confirm HPRT deficiency?
A. Enzyme assay in erythrocytes
B. Glucose tolerance test
C. Liver function test
D. Serum amylase -
In Lesch–Nyhan Syndrome, PRPP levels are:
A. Decreased
B. Increased
C. Normal
D. Undetectable -
Which of the following neurotransmitters is found decreased in Lesch–Nyhan Syndrome?
A. Dopamine
B. Serotonin
C. GABA
D. Acetylcholine -
Which of the following best describes the age of onset?
A. At birth
B. After puberty
C. During infancy (3–6 months)
D. After 10 years of age -
Carrier females can be identified by:
A. Skin biopsy
B. HPRT enzyme assay or DNA analysis
C. Urine uric acid
D. MRI scan -
The neurological symptoms of Lesch–Nyhan Syndrome are due to dysfunction in which neurotransmitter pathway?
A. Dopaminergic
B. Cholinergic
C. GABAergic
D. Serotonergic -
Which of the following laboratory findings is most characteristic?
A. Low uric acid
B. High uric acid
C. Low ammonia
D. High bilirubin -
Which of the following is a diagnostic imaging finding in Lesch–Nyhan Syndrome?
A. Atrophy of basal ganglia
B. Enlargement of ventricles
C. Calcification of cerebellum
D. Brainstem tumor -
Which therapy primarily targets uric acid reduction?
A. Levodopa
B. Allopurinol
C. Diazepam
D. Baclofen -
What is the main limitation of allopurinol therapy in LNS?
A. It causes hypouricemia
B. It doesn’t improve neurological symptoms
C. It increases uric acid excretion
D. It is toxic to kidneys -
Life expectancy in untreated Lesch–Nyhan Syndrome is usually:
A. Less than 10 years
B. 10–15 years
C. 20–30 years with supportive care
D. 50–60 years
Answer Key
-
C — Hypoxanthine-guanine phosphoribosyltransferase
-
C — X-linked recessive
-
B — Only males
-
C — Chromosome X (Xq26–Xq27)
-
C — Hyperuricemia
-
B — Purine salvage pathway
-
B — HPRT
-
B — Dystonia and spasticity
-
B — Dopamine
-
B — Sodium urate
-
C — Hypouricemia
-
B — Hyperuricemia, neurological dysfunction, self-mutilation
-
B — Lesch and Nyhan
-
B — Inhibit uric acid synthesis
-
A — Xanthine oxidase
-
C — Both A and B
-
B — Basal ganglia
-
A — Autophagia
-
B — Increased de novo purine synthesis
-
A — Enzyme assay in erythrocytes
-
B — Increased
-
A — Dopamine
-
C — During infancy (3–6 months)
-
B — HPRT enzyme assay or DNA analysis
-
A — Dopaminergic
-
B — High uric acid
-
A — Atrophy of basal ganglia
-
B — Allopurinol
-
B — It doesn’t improve neurological symptoms
-
C — 20–30 years with supportive care