Introduction
- Beta-oxidation of fatty acids is the metabolic pathway by which fatty acids are broken down in the mitochondria (and peroxisomes) of cells to generate energy.
- This process involves the sequential removal of two-carbon units from the fatty acid chain in the form of acetyl-CoA.
- The term “beta-oxidation” comes from the fact that oxidation occurs at the beta carbon (the third carbon) of the fatty acid chain.
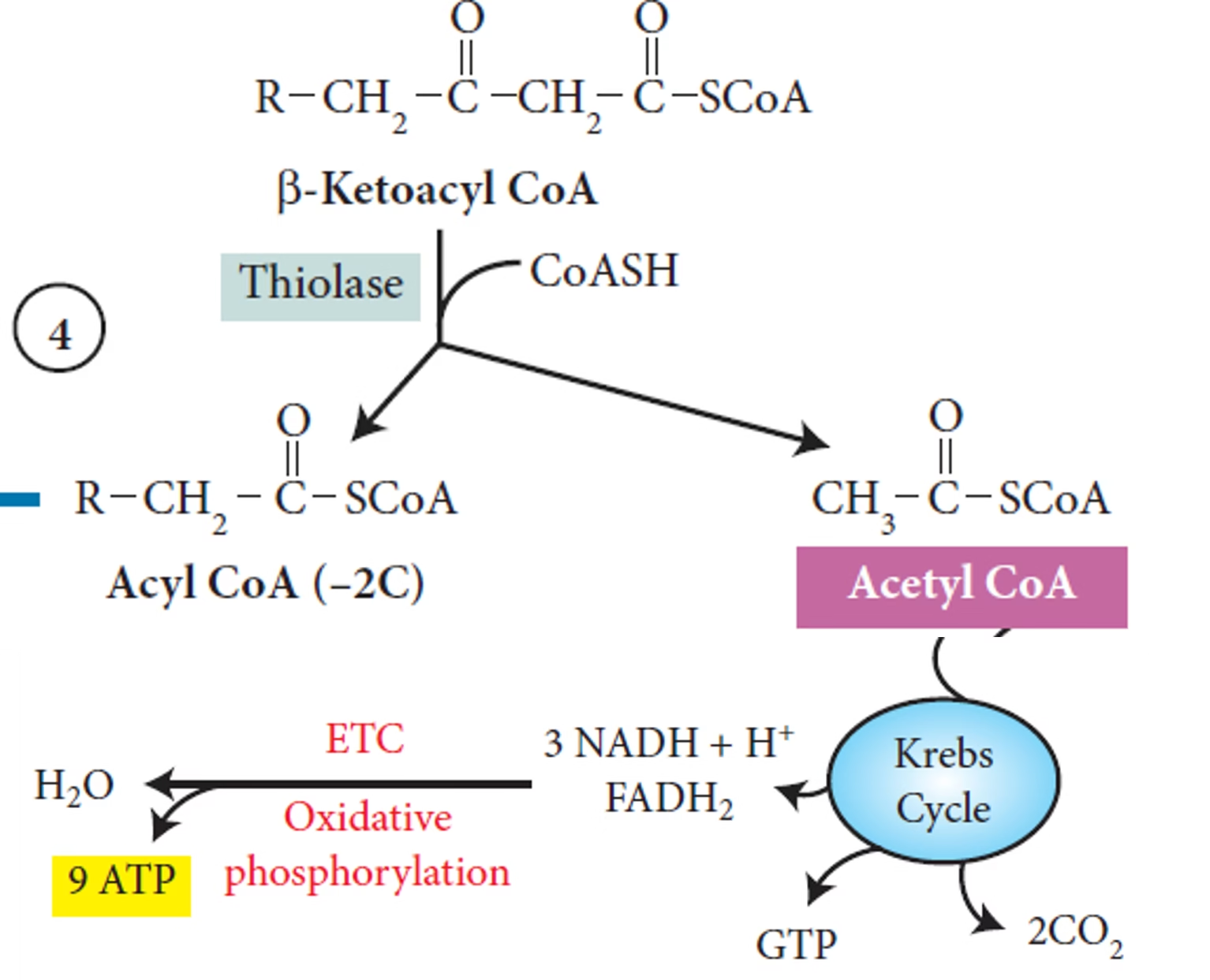
- The acetyl-CoA produced enters the citric acid cycle (Krebs cycle), while NADH and FADH₂ generated during the process enter the electron transport chain to produce ATP, the energy currency of the cell.
- Beta-oxidation plays a crucial role during fasting, prolonged exercise, and starvation, when the body relies on fat stores for energy instead of glucose.
Role of Fatty Acids in Metabolism
Fatty Acids as a Primary Energy Source
🔸 High Energy Yield
-
Fatty acids are highly reduced hydrocarbons; thus, their oxidation yields significantly more ATP compared to carbohydrates and amino acids.
-
For instance, the complete mitochondrial β-oxidation of palmitate (C16:0) yields:
-
8 Acetyl-CoA → TCA cycle → 80 ATP (via NADH, FADH₂)
-
7 NADH → 17.5 ATP
-
7 FADH₂ → 10.5 ATP
-
Net yield: ~106 ATP
-
🔸 Tissue Specific Utilization
-
Liver, skeletal muscle, cardiac muscle, and kidneys rely heavily on FA oxidation during fasting states.
-
The brain cannot utilize fatty acids directly due to the blood-brain barrier, but can utilise ketone bodies derived from fatty acid metabolism.
Lipid Storage and Mobilization
🔸 Triglyceride Storage
-
Fatty acids are esterified with glycerol to form triacylglycerols (TAGs) and stored in adipose tissue.
-
TAGs represent the most concentrated form of energy storage in the human body.
🔸 Lipolysis and FA Transport
-
During fasting, hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL) are activated by glucagon and epinephrine, catalyzing TAG hydrolysis.
-
Free fatty acids are released into circulation and transported bound to albumin.
Mitochondrial β-Oxidation
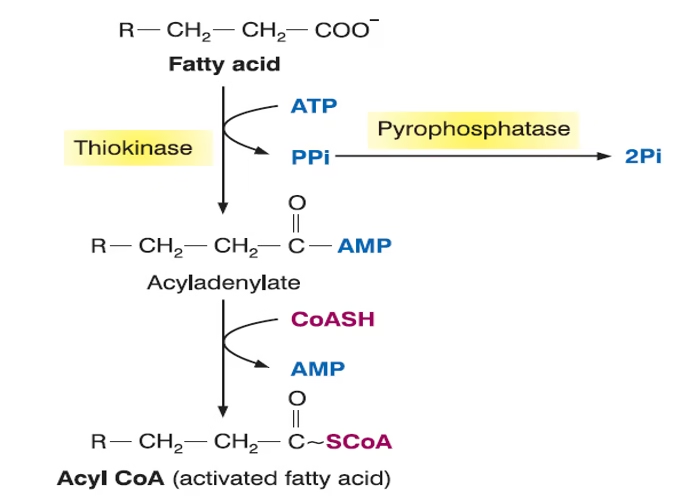
🔸 Activation and Transport
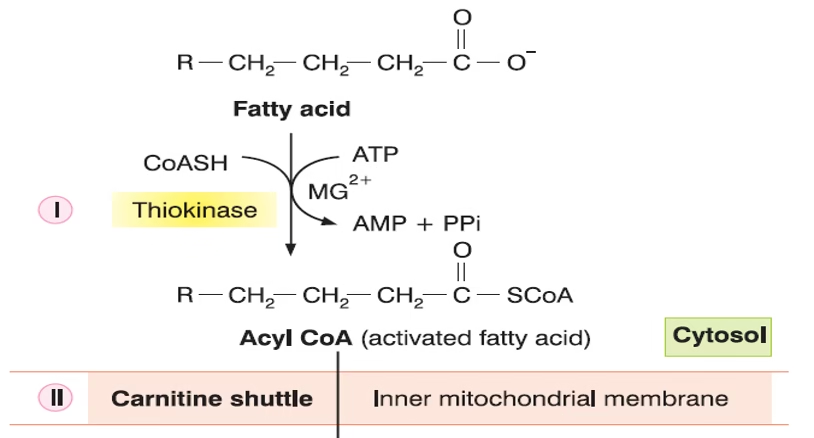
-
FA is activated to fatty acyl-CoA in the cytosol (via acyl-CoA synthetase).
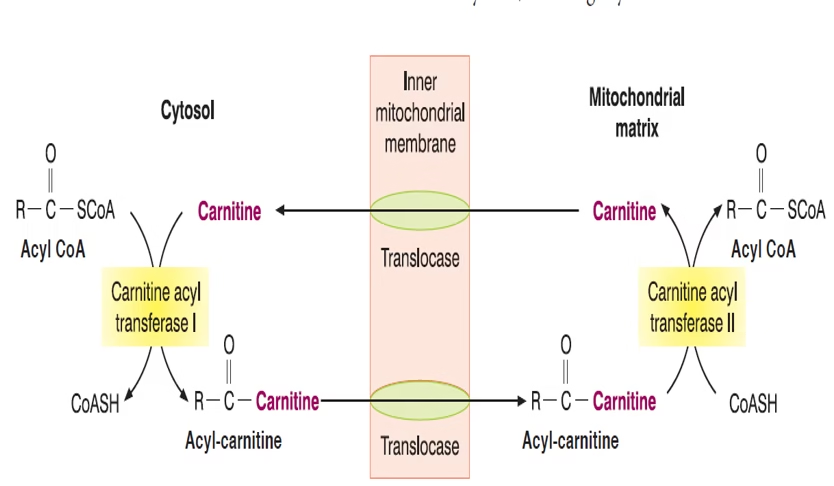
-
Transported into mitochondria via the carnitine shuttle (CPT-I, translocase, CPT-II).
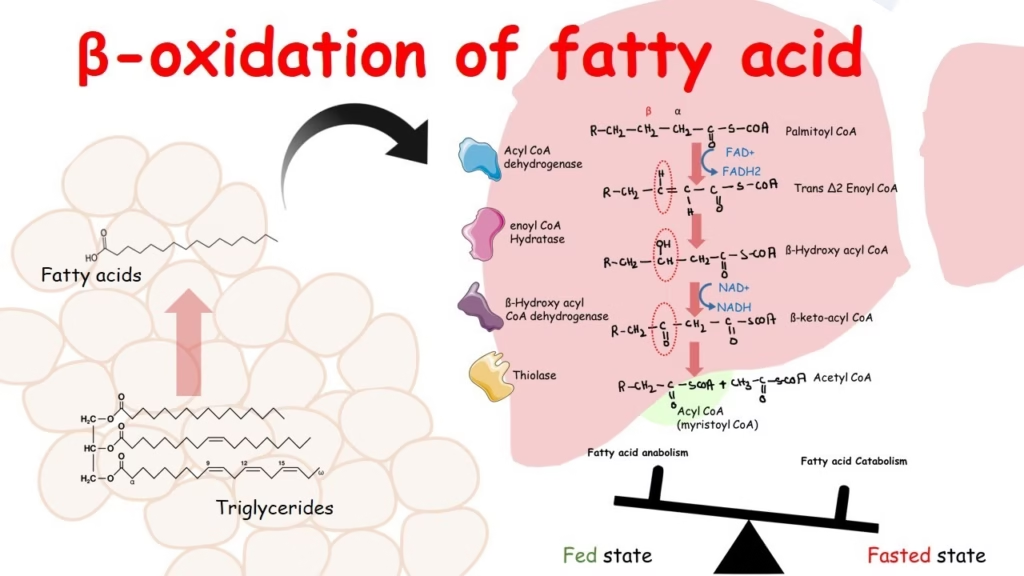
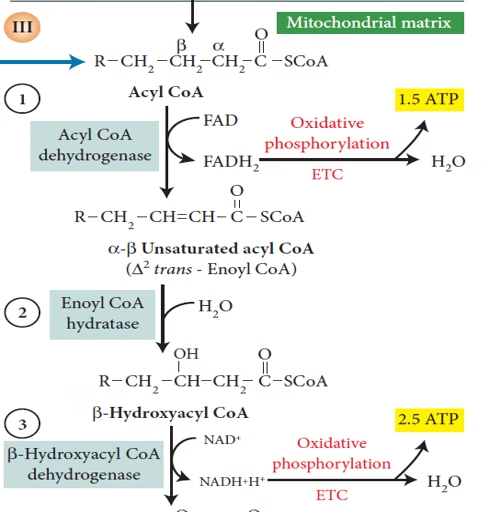
🔸 Sequential Oxidation
Each β-oxidation cycle includes:
-
Dehydrogenation (acyl-CoA dehydrogenase) → FADH₂
-
Hydration (enoyl-CoA hydratase)
-
Dehydrogenation (L-3-hydroxyacyl-CoA dehydrogenase) → NADH
-
Thiolysis (β-ketothiolase) → Acetyl-CoA
Ketogenesis: Adaptation to Glucose Sparing
-
In prolonged fasting, hepatic β-oxidation provides abundant acetyl-CoA, exceeding the TCA cycle capacity.
-
Acetyl-CoA is diverted to synthesize ketone bodies (acetoacetate, β-hydroxybutyrate, acetone).
-
Ketone bodies act as an alternative fuel for brain, heart, and skeletal muscles.
Fatty Acids as Signalling Molecules
-
Fatty acids modulate cell signalling by acting as:
-
Ligands for PPARs (Peroxisome Proliferator-Activated Receptors): regulate lipid metabolism, insulin sensitivity, and inflammation.
-
Precursors for eicosanoids (e.g., prostaglandins, leukotrienes, thromboxanes): derived from arachidonic acid, regulate immunity, vasodilation, and clotting.
-
Structural Role in Membranes
-
Fatty acids are integral to phospholipids and sphingolipids in cellular membranes.
-
Influence membrane fluidity, permeability, and lipid raft formation.
-
PUFAs (polyunsaturated fatty acids), especially omega-3 and omega-6, are vital for neural membranes and retinal function.
Biosynthesis and Anabolic Functions
-
De novo fatty acid synthesis occurs in the cytosol of liver and adipose tissue.
-
Enzyme: fatty acid synthase (FAS) complex elongates acetyl-CoA and malonyl-CoA to form palmitate.
-
NADPH from pentose phosphate pathway is required.
Fatty Acid Oxidation
The β-oxidation of fatty acids occurs in three stages.
- Activation of fatty acids in the cytosol
- Transport of fatty acids from the cytosol to the mitochondria
- Reactions of β-oxidation in the mitochondrial matrix.
Activation of fatty acids in the cytosol
Transport of fatty acids from the cytosol to the mitochondria
Reactions of β-oxidation in the mitochondrial matrix.
Clinical Aspects
Inherited Disorders
These are a group of rare, genetic metabolic disorders caused by mutations in enzymes involved in beta-oxidation. Common examples include:
-
MCAD Deficiency (Medium-Chain Acyl-CoA Dehydrogenase Deficiency):
-
Most common FAOD.
-
Symptoms: Hypoglycemia, vomiting, lethargy, seizures, sudden death.
-
Triggered by fasting or infections.
-
-
LCHAD Deficiency (Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency):
-
Can lead to liver dysfunction, cardiomyopathy, and rhabdomyolysis.
-
Seen in infancy or early childhood.
-
-
CPT-I and CPT-II Deficiency (Carnitine Palmitoyltransferase Deficiencies):
-
Affects transport of long-chain fatty acids into mitochondria.
-
CPT-I: Presents with hypoketotic hypoglycemia.
-
CPT-II: Muscle weakness, myoglobinuria after prolonged exercise.
-
Clinical Features of FAODs
-
Fasting intolerance
-
Hypoketotic hypoglycemia (low blood sugar with low ketone bodies)
-
Liver dysfunction (hepatomegaly, elevated liver enzymes)
-
Cardiomyopathy (especially in long-chain FAODs)
-
Muscle weakness and rhabdomyolysis
-
Sudden infant death (SIDS) association in undiagnosed cases
Diagnosis
-
Blood and urine tests: Hypoglycemia without ketones, elevated liver enzymes, abnormal acylcarnitine profile.
-
Newborn screening: Tandem mass spectrometry.
-
Genetic testing: Identification of mutations.
-
Enzyme assay: To assess specific enzyme deficiencies.