
Introduction
-
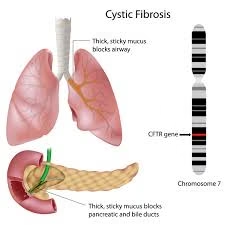
Cystic fibrosis (CF) is a hereditary, autosomal recessive disorder.
-
It primarily affects the respiratory, digestive, and sweat gland systems.
-
The disease is caused by mutations in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene.
-
CFTR gene is located on chromosome 7.
-
Defective CFTR protein leads to abnormal chloride and sodium transport across epithelial cells.
-
This results in the production of thick, sticky mucus.
-
The thick mucus causes obstruction of airways and ducts, leading to chronic infections and inflammation.
-
Cystic fibrosis is one of the most common life-limiting genetic disorders in children.
-
It affects multiple organs including lungs, pancreas, liver, intestines, and reproductive organs.
-
Early diagnosis and appropriate management have significantly improved survival and quality of life.
Etiology and Genetics
-
Cystic fibrosis is caused by mutations in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene
-
The CFTR gene is located on chromosome 7
-
Inheritance pattern: Autosomal recessive
-
Both parents must be carriers for the child to be affected
Common Mutation
-
ΔF508 (Delta F508) mutation is the most common
-
Leads to misfolding and degradation of CFTR protein
Pathophysiology
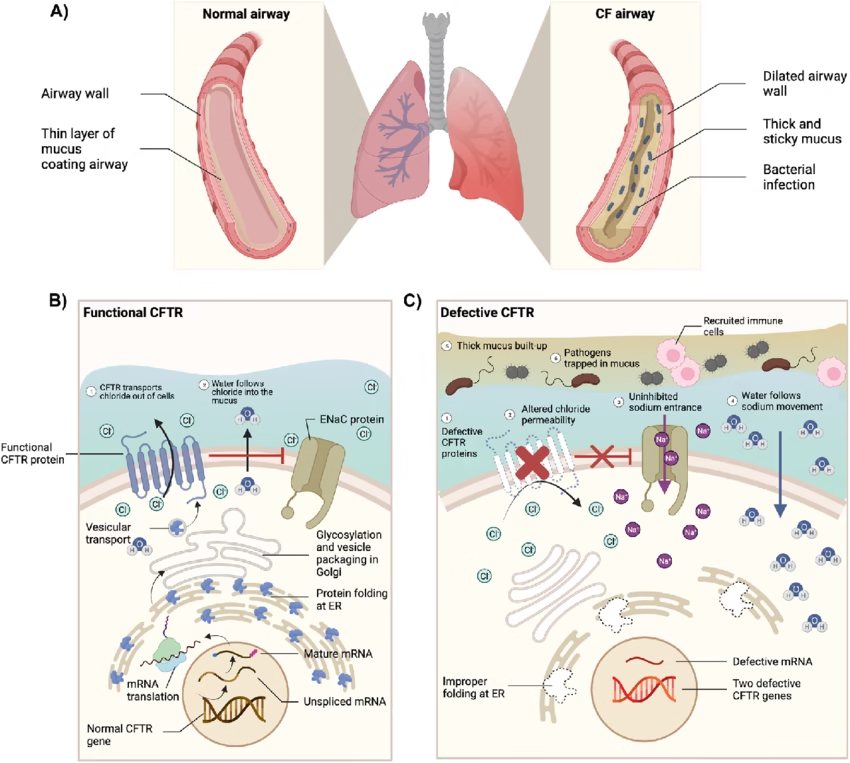
The CFTR protein functions as a chloride ion channel in epithelial cells.
Normal Function
-
Regulates chloride and sodium transport
-
Maintains hydration of secretions
In Cystic Fibrosis
-
Defective chloride secretion
-
Increased sodium and water reabsorption
-
Dehydrated, thick mucus
Consequences
-
Airway obstruction
-
Chronic bacterial infections
-
Pancreatic duct blockage
-
Malabsorption
Organ System Involvement
1. Respiratory System
-
Thick mucus blocks the bronchi and bronchioles
-
Chronic cough with sputum
-
Recurrent lung infections (Pseudomonas, Staphylococcus)
-
Bronchiectasis
-
Progressive respiratory failure (leading cause of death)
2. Digestive System
-
Pancreatic duct obstruction
-
Deficiency of digestive enzymes
-
Fat malabsorption
-
Steatorrhea
-
Failure to thrive in children
-
Deficiency of fat-soluble vitamins (A, D, E, K)
3. Sweat Glands
-
Increased chloride and sodium in sweat
-
Basis of diagnostic sweat test
-
Risk of dehydration and electrolyte imbalance
4. Reproductive System
-
Male infertility due to congenital absence of vas deferens
-
Reduced fertility in females due to thick cervical mucus
Clinical Features
-
Chronic productive cough
-
Recurrent chest infections
-
Failure to thrive
-
Bulky, greasy stools
-
Salty taste of skin
-
Nasal polyps
-
Digital clubbing (late stage)
Diagnosis
1. Sweat Chloride Test (Gold Standard)
-
Measures chloride concentration in sweat
-
Based on defective CFTR-mediated chloride reabsorption
-
Chloride ≥ 60 mmol/L → diagnostic of cystic fibrosis
-
30–59 mmol/L → intermediate (requires repeat testing)
-
< 30 mmol/L → unlikely CF
-
Performed using pilocarpine iontophoresis
2. Newborn Screening
-
Initial screening test in many countries
-
Based on elevated immunoreactive trypsinogen (IRT) in blood
-
Positive screening requires confirmatory sweat test or genetic testing
-
Helps in early diagnosis before symptoms appear
3. Genetic Testing
-
Detects CFTR gene mutations
-
Useful for:
-
Confirming diagnosis
-
Identifying carriers
-
Prenatal diagnosis
-
-
Common mutation detected: ΔF508
4. Pulmonary Function Tests (PFTs)
-
Assess lung involvement and disease severity
-
Shows:
-
Obstructive pattern
-
Reduced FEV₁
-
-
Used for monitoring progression, not primary diagnosis
5. Microbiological Examination
-
Sputum or throat swab culture
-
Identifies chronic infections like:
-
Pseudomonas aeruginosa
-
Staphylococcus aureus
-
-
Guides antibiotic therapy
6. Imaging Studies
-
Chest X-ray / HRCT chest
-
Findings include:
-
Hyperinflation
-
Bronchiectasis
-
Mucus plugging
-
-
Helpful in assessing extent of lung damage
7. Pancreatic Function Tests
-
Low fecal elastase
-
Evidence of exocrine pancreatic insufficiency
-
Supports diagnosis in symptomatic patients
Management
Cystic fibrosis has no cure, but early and aggressive management improves survival.
Respiratory Management
-
Chest physiotherapy
-
Mucolytics (e.g., DNase)
-
Bronchodilators
-
Antibiotics for infections
Nutritional Management
-
Pancreatic enzyme replacement therapy
-
High-calorie, high-protein diet
-
Supplementation of fat-soluble vitamins
CFTR Modulator Therapy
-
Target specific CFTR mutations
-
Improves CFTR function
-
Example: correctors and potentiators
Complications
-
Chronic respiratory failure
-
Cor pulmonale
-
Diabetes mellitus (CFRD)
-
Liver disease
-
Osteoporosis
Prognosis
-
Prognosis has significantly improved with early diagnosis
-
Median survival now exceeds 40–50 years in many countries
-
Quality of life depends on early treatment and compliance
Prevention and Genetic Counseling
-
Carrier screening for at-risk couples
-
Prenatal diagnosis
-
Genetic counseling is essential
MCQs
1. Cystic fibrosis is inherited as:
A. Autosomal dominant
B. X-linked recessive
C. Autosomal recessive
D. Mitochondrial
2. The gene defective in cystic fibrosis is:
A. BRCA1
B. CFTR
C. HBB
D. p53
3. CFTR gene is located on which chromosome?
A. Chromosome 5
B. Chromosome 6
C. Chromosome 7
D. Chromosome 11
4. CFTR protein primarily functions as a:
A. Sodium channel
B. Potassium channel
C. Chloride channel
D. Calcium pump
5. Most common mutation in cystic fibrosis is:
A. G551D
B. R117H
C. ΔF508
D. N1303K
6. Pathophysiology of CF mainly results in:
A. Increased water secretion
B. Thick, dehydrated mucus
C. Excess mucus clearance
D. Alkaline secretions
7. The organ system most commonly affected in CF is:
A. Nervous system
B. Cardiovascular system
C. Respiratory system
D. Endocrine system
8. Major cause of morbidity and mortality in CF patients is:
A. Liver failure
B. Renal failure
C. Respiratory failure
D. Cardiac arrhythmia
9. Thick mucus in CF leads to:
A. Improved gas exchange
B. Airway obstruction
C. Increased ciliary movement
D. Reduced infections
10. Common organism causing lung infection in CF is:
A. Mycobacterium tuberculosis
B. Pseudomonas aeruginosa
C. Streptococcus pyogenes
D. Klebsiella pneumoniae
11. Pancreatic involvement in CF causes:
A. Hypersecretion of insulin
B. Exocrine pancreatic insufficiency
C. Acute pancreatitis
D. Pancreatic carcinoma
12. Fat malabsorption in CF leads to deficiency of:
A. Water-soluble vitamins
B. Vitamin B complex
C. Fat-soluble vitamins
D. Vitamin C
13. Steatorrhea in CF occurs due to deficiency of:
A. Bile salts
B. Digestive enzymes
C. Gastric acid
D. Intestinal hormones
14. Sweat chloride test is based on abnormal:
A. Sodium secretion
B. Potassium secretion
C. Chloride reabsorption
D. Calcium absorption
15. Diagnostic sweat chloride value for CF is:
A. < 30 mmol/L
B. 30–40 mmol/L
C. > 60 mmol/L
D. > 100 mmol/L only
16. Gold standard diagnostic test for CF is:
A. Chest X-ray
B. Genetic testing
C. Sweat chloride test
D. Pulmonary function test
17. Newborn screening for CF commonly measures:
A. Serum amylase
B. Immunoreactive trypsinogen (IRT)
C. Sweat sodium
D. Serum lipase
18. CFTR dysfunction causes increased reabsorption of:
A. Chloride
B. Potassium
C. Sodium and water
D. Calcium
19. Lung pathology in CF commonly shows:
A. Emphysema
B. Bronchiectasis
C. Pleural effusion
D. Pneumothorax only
20. Male infertility in CF is due to:
A. Testicular atrophy
B. Low testosterone
C. Absence of vas deferens
D. Erectile dysfunction
21. Female infertility in CF is mainly due to:
A. Ovarian failure
B. Thick cervical mucus
C. Hormonal imbalance
D. Endometrial damage
22. CF-related diabetes occurs due to:
A. Autoimmune destruction
B. Insulin resistance only
C. Pancreatic damage
D. Excess insulin
23. Which vitamin deficiency is common in CF?
A. Vitamin C
B. Vitamin A
C. Vitamin B12
D. Vitamin B1
24. Chest physiotherapy in CF helps in:
A. Reducing inflammation
B. Improving nutrition
C. Clearing mucus
D. Treating infection
25. Dornase alfa acts by:
A. Breaking DNA in mucus
B. Killing bacteria
C. Dilating bronchi
D. Reducing inflammation
26. CFTR modulators are used to:
A. Cure CF completely
B. Treat infections only
C. Improve CFTR protein function
D. Replace pancreatic enzymes
27. CFTR modulators are mutation-specific because:
A. All mutations behave similarly
B. CFTR protein defects vary
C. They act on bacteria
D. They suppress immunity
28. High sweat chloride in CF results in:
A. Hypotension
B. Electrolyte imbalance
C. Metabolic alkalosis
D. Hyperglycemia
29. Clubbing of fingers in CF indicates:
A. Early disease
B. Acute infection
C. Chronic hypoxia
D. Cardiac failure
30. CF is most common in which population?
A. African
B. Asian
C. Caucasian
D. Native American
31. Mode of inheritance requires parents to be:
A. Both affected
B. One affected
C. Both carriers
D. One carrier only
32. Fecal elastase test assesses:
A. Liver function
B. Intestinal absorption
C. Pancreatic exocrine function
D. Renal function
33. Major cause of failure to thrive in CF children is:
A. Poor appetite
B. Chronic fever
C. Malabsorption
D. Anemia
34. Chronic infection in CF lungs leads to:
A. Reduced inflammation
B. Fibrosis and bronchiectasis
C. Alveolar collapse only
D. Pleural thickening
35. Which electrolyte is increased in CF sweat?
A. Calcium
B. Magnesium
C. Chloride
D. Bicarbonate
36. Prenatal diagnosis of CF is done by:
A. Ultrasound
B. Sweat test
C. Genetic testing
D. Chest X-ray
37. CF patients are advised high-calorie diet because of:
A. Increased appetite
B. Increased metabolic demand
C. Reduced physical activity
D. Hormonal imbalance
38. Which enzyme replacement is used in CF?
A. Lactase
B. Pepsin
C. Pancreatic enzymes
D. Amylase only
39. CF primarily affects epithelial cells of:
A. Blood vessels
B. Muscle
C. Exocrine glands
D. Neurons
40. Primary defect in CF is in transport of:
A. Proteins
B. Lipids
C. Electrolytes
D. Glucose
41. CF lung disease is characterized by:
A. Dry cough
B. Productive cough
C. No cough
D. Hemoptysis only
42. Chronic hypoxia in CF may cause:
A. Polycythemia
B. Leukopenia
C. Thrombocytopenia
D. Anemia
43. CFTR protein belongs to which family?
A. G-protein receptors
B. Ion channels
C. ABC transporter family
D. Nuclear receptors
44. Which test helps monitor lung disease progression?
A. Sweat test
B. Genetic test
C. Pulmonary function test
D. Stool examination
45. Main aim of CF management is to:
A. Cure disease
B. Prevent complications
C. Improve quality of life
D. Both B and C
46. CF patients commonly have stools that are:
A. Hard and dry
B. Watery
C. Bulky and greasy
D. Bloody
47. Liver involvement in CF may lead to:
A. Cirrhosis
B. Hepatitis A
C. Fatty liver only
D. Liver cancer
48. Early diagnosis of CF improves:
A. Cure rate
B. Survival and quality of life
C. Genetic mutation
D. Sweat chloride levels
49. CF is a life-limiting disease mainly because of:
A. Pancreatic failure
B. Renal failure
C. Progressive lung disease
D. Liver failure
50. Genetic counseling in CF is important for:
A. Treating disease
B. Carrier detection
C. Preventing inheritance
D. All of the above
Answer Key
-
C
-
B
-
C
-
C
-
C
-
B
-
C
-
C
-
B
-
B
-
B
-
C
-
B
-
C
-
C
-
C
-
B
-
C
-
B
-
C
-
B
-
C
-
B
-
C
-
A
-
C
-
B
-
B
-
C
-
C
-
C
-
C
-
C
-
B
-
C
-
C
-
B
-
C
-
C
-
C
-
B
-
A
-
C
-
C
-
D
-
C
-
A
-
B
-
C
-
D