
Introduction
-
Hemoglobin is a complex iron-containing protein present in red blood cells.
-
It is responsible for the transport of oxygen from the lungs to body tissues.
-
Hemoglobin also plays a crucial role in transporting carbon dioxide from tissues to the lungs.
-
Each hemoglobin molecule consists of four globin chains and four heme groups.
-
The iron present in the heme group enables reversible binding of oxygen.
-
Hemoglobin plays a significant role in maintaining blood pH by acting as a buffer.
-
It is synthesized in the erythroid precursor cells of the bone marrow.
-
Normal hemoglobin structure and function are essential for efficient tissue oxygenation.
-
Abnormalities in hemoglobin synthesis or structure result in anemia and hemoglobinopathies.
-
Understanding hemoglobin is fundamental in biochemistry, hematology, and clinical diagnosis.
Hemoglobin Synthesis
Hemoglobin synthesis occurs mainly in the erythroid precursor cells of the bone marrow and involves the coordinated production of globin chains and heme, which later assemble to form a functional hemoglobin molecule.

Haemoglobin comprises four polypeptide chains, each containing an iron-bound heme group. The synthesis of Hemoglobin involves several steps:
Hemoglobin Structure
-
Hemoglobin is a tetrameric conjugated protein present inside red blood cells.
-
Each hemoglobin molecule consists of four polypeptide (globin) chains.
-
In adult hemoglobin (HbA), the globin chains are:
-
Two alpha (α) chains
-
Two beta (β) chains
-
-
Each globin chain is tightly associated with a heme group, making a total of four heme groups per hemoglobin molecule.
-
The heme group is a non-protein prosthetic group composed of:
-
A porphyrin ring (protoporphyrin IX)
-
A centrally placed ferrous iron (Fe²⁺) atom
-
-
The iron atom forms reversible bonds with oxygen, allowing hemoglobin to bind and release oxygen efficiently.
-
Each heme group can bind one molecule of oxygen, so one hemoglobin molecule can carry four oxygen molecules.
-
The globin chains are arranged in a quaternary structure, which is essential for cooperative oxygen binding.
-
The interaction between globin chains allows hemoglobin to shift between:
-
Tense (T) state – low oxygen affinity
-
Relaxed (R) state – high oxygen affinity
-
-
This unique structural arrangement makes hemoglobin an efficient oxygen transporter under varying physiological conditions.

Synthesis of Globin Chains
Chromosomal Location of Globin Genes
-
Alpha (α) globin genes → Chromosome 16
-
Beta (β), Gamma (γ), Delta (δ) globin genes → Chromosome 11
Types of Globin Chains and Their Expression
-
α chains: Synthesized throughout fetal and adult life
-
γ chains: Predominant during fetal life → form HbF
-
β chains: Predominant after birth → form HbA
-
δ chains: Minor component in adults → form HbA₂
This transition from γ to β chain synthesis after birth is known as globin gene switching.
Steps in Synthesis of Globin Chains
1. Transcription
-
Occurs in the nucleus of erythroid precursor cells.
-
Globin genes are transcribed into messenger RNA (mRNA).
-
Regulated by transcription factors and regulatory DNA sequences.
2. mRNA Processing
-
Newly formed mRNA undergoes:
-
Capping
-
Polyadenylation
-
Removal of introns (splicing)
-
-
Mature mRNA exits the nucleus into the cytoplasm.
3. Translation
-
Takes place on ribosomes in the cytoplasm.
-
mRNA is translated into globin polypeptide chains.
-
Each chain folds into its specific secondary and tertiary structure.
4. Post-Translational Stability
-
Globin chains are unstable without heme.
-
Binding of heme protects globin chains from degradation.
-
Balanced synthesis ensures efficient hemoglobin assembly.
Regulation of Globin Chain Synthesis
-
Controlled by gene availability, transcription rate, and mRNA stability.
-
Heme availability enhances globin chain synthesis.
-
Erythropoietin indirectly stimulates globin production by increasing erythropoiesis.
-
Imbalance between α and β chain synthesis leads to pathological conditions.
Table: Summary of Globin Chain Synthesis
| Globin Chain | Chromosome | Period of Predominance | Hemoglobin Formed | Clinical Significance |
|---|---|---|---|---|
| α (Alpha) | 16 | Fetal & Adult | HbF, HbA, HbA₂ | Defect → α-thalassemia |
| β (Beta) | 11 | Adult | HbA | Defect → β-thalassemia |
| γ (Gamma) | 11 | Fetal life | HbF | Higher O₂ affinity |
| δ (Delta) | 11 | Adult (minor) | HbA₂ | Raised in β-thalassemia |
Synthesis of Heme
Cellular Location of Heme Synthesis
-
Mitochondria: Initial and final steps
-
Cytosol: Intermediate steps
-
This compartmentalization is crucial for regulation and efficient synthesis.
Steps of Heme Synthesis
Step 1: Formation of δ-Aminolevulinic Acid (ALA)
-
Occurs in the mitochondria
-
Glycine + Succinyl-CoA → δ-ALA
-
Enzyme: ALA synthase (rate-limiting enzyme)
-
Requires pyridoxal phosphate (vitamin B₆)
-
This step is inhibited by heme (feedback inhibition)
Step 2: Formation of Porphobilinogen
-
Occurs in the cytosol
-
2 ALA → Porphobilinogen
-
Enzyme: ALA dehydratase
-
Highly sensitive to lead inhibition
Step 3: Formation of Hydroxymethylbilane
-
4 Porphobilinogen → Hydroxymethylbilane
-
Enzyme: Porphobilinogen deaminase
-
Defect leads to acute intermittent porphyria
Step 4: Formation of Uroporphyrinogen III
-
Hydroxymethylbilane is cyclized to uroporphyrinogen III
-
Enzyme: Uroporphyrinogen III synthase
Step 5: Formation of Coproporphyrinogen III
-
Occurs in cytosol
-
Uroporphyrinogen III undergoes decarboxylation
-
Enzyme: Uroporphyrinogen decarboxylase
Step 6: Formation of Protoporphyrin IX
-
Coproporphyrinogen III re-enters mitochondria
-
Converted to protoporphyrin IX
-
Enzymes: Coproporphyrinogen oxidase and Protoporphyrinogen oxidase
Step 7: Formation of Heme
-
Final mitochondrial step
-
Protoporphyrin IX + Fe²⁺ → Heme
-
Enzyme: Ferrochelatase
-
Also inhibited by lead
Table: Steps of Heme Synthesis
| Step | Reaction | Enzyme | Location | Clinical Importance |
|---|---|---|---|---|
| 1 | Glycine + Succinyl-CoA → ALA | ALA synthase | Mitochondria | Rate-limiting; needs Vit B₆ |
| 2 | ALA → Porphobilinogen | ALA dehydratase | Cytosol | Inhibited by lead |
| 3 | Porphobilinogen → Hydroxymethylbilane | PBG deaminase | Cytosol | Defect → AIP |
| 4 | Hydroxymethylbilane → Uroporphyrinogen III | Uroporphyrinogen III synthase | Cytosol | Prevents abnormal porphyrins |
| 5 | Uroporphyrinogen III → Coproporphyrinogen III | Uroporphyrinogen decarboxylase | Cytosol | Defect → Porphyria cutanea tarda |
| 6 | Coproporphyrinogen III → Protoporphyrin IX | Oxidases | Mitochondria | Builds porphyrin ring |
| 7 | Protoporphyrin IX + Fe²⁺ → Heme | Ferrochelatase | Mitochondria | Inhibited by lead |
Regulation of Heme Synthesis
-
ALA synthase is the key regulatory enzyme.
-
Heme acts as a feedback inhibitor of ALA synthase.
-
Iron availability directly influences the final step of heme formation.
-
In erythroid cells, synthesis is regulated by globin chain availability.
Hemoglobin Assembly
1. Association of Heme with Globin Chains
-
Each synthesized globin chain binds one heme molecule.
-
This binding occurs mainly in the cytoplasm of erythroid cells.
-
The iron (Fe²⁺) of heme forms a strong coordination bond with the globin chain.
-
Proper heme insertion is essential for globin chain stability.
2. Formation of Globin–Heme Monomers
-
A single globin chain plus one heme group forms a heme–globin monomer.
-
This step ensures that globin chains are protected from degradation.
-
Free globin chains without heme are unstable and rapidly degraded.
3. Dimer Formation
-
One α-globin–heme complex pairs with one β-globin–heme complex to form an αβ dimer.
-
In fetal life, α-globin pairs with γ-globin to form αγ dimers.
-
Dimer formation is a prerequisite for tetramer assembly.
4. Tetramer Formation
-
Two αβ (or αγ) dimers associate to form a hemoglobin tetramer.
-
Adult hemoglobin: α₂β₂ (HbA)
-
Fetal hemoglobin: α₂γ₂ (HbF)
-
The tetrameric structure is stabilized by non-covalent interactions.
5. Development of Functional Conformation
-
The tetramer exists in two interconvertible states:
-
Tense (T) state – low oxygen affinity
-
Relaxed (R) state – high oxygen affinity
-
-
Binding of oxygen shifts hemoglobin from T-state to R-state.
-
This structural transition is responsible for cooperative oxygen binding.
Factors Influencing Hemoglobin Assembly
-
Balanced globin chain synthesis is essential for effective assembly.
-
Availability of iron and intact heme synthesis pathway.
-
Adequate erythropoietin stimulation for erythropoiesis.
-
Absence of genetic defects in globin genes.
Table: Hemoglobin Assembly
| Stage | Event | Key Feature | Clinical Relevance |
|---|---|---|---|
| 1 | Heme binds globin | Stabilizes globin chains | Defect → ineffective erythropoiesis |
| 2 | Monomer formation | One globin + one heme | Prevents globin degradation |
| 3 | Dimer formation | αβ or αγ pairing | Required for tetramer |
| 4 | Tetramer formation | α₂β₂ or α₂γ₂ | Functional hemoglobin |
| 5 | Conformational change | T ↔ R state | Enables oxygen delivery |
Hemoglobin Function
1. Oxygen Transport
-
Hemoglobin transports oxygen (O₂) from the lungs to peripheral tissues.
-
In the lungs, where partial pressure of oxygen (pO₂) is high, oxygen binds to hemoglobin forming oxyhemoglobin.
-
Each hemoglobin molecule can carry four oxygen molecules, one per heme group.
-
In tissues with low pO₂, hemoglobin releases oxygen for cellular metabolism.
-
This process is facilitated by the sigmoidal oxygen–hemoglobin dissociation curve, indicating cooperative binding.
Physiological Advantages
-
Ensures efficient oxygen loading in lungs.
-
Enables maximal oxygen unloading in metabolically active tissues.
2. Carbon Dioxide Transport
-
Hemoglobin helps transport carbon dioxide (CO₂) from tissues to the lungs.
-
CO₂ binds to the globin portion of hemoglobin forming carbaminohemoglobin.
-
Approximately 20–30% of CO₂ is transported in this form.
-
Deoxygenated hemoglobin binds CO₂ more readily (Haldane effect).
3. Acid–Base Buffering Action
-
Hemoglobin acts as a powerful buffer system in blood.
-
It binds hydrogen ions (H⁺) released during tissue metabolism.
-
This buffering capacity helps maintain normal blood pH (≈7.4).
-
Hemoglobin works in coordination with the bicarbonate buffer system.
4. Transport of Nitric Oxide (NO)
-
Hemoglobin modulates nitric oxide availability, influencing vascular tone.
-
NO binding contributes to vasodilation, improving tissue perfusion.
5. Regulation of Oxygen Delivery (Bohr Effect)
-
Increased CO₂, H⁺ concentration, and temperature reduce hemoglobin’s oxygen affinity.
-
This promotes oxygen release in actively metabolizing tissues.
-
Known as the Bohr effect, it enhances tissue oxygenation.
Table: Functions of Hemoglobin
| Function | Mechanism | Physiological Significance |
|---|---|---|
| Oxygen transport | Reversible O₂ binding to heme iron | Tissue oxygenation |
| CO₂ transport | Carbaminohemoglobin formation | Removal of metabolic waste |
| Buffering action | Binding of H⁺ ions | Maintains blood pH |
| NO transport | Modulation of NO availability | Regulates blood flow |
| Bohr effect | Reduced O₂ affinity at low pH/high CO₂ | Enhanced O₂ release |
Clinical Importance
-
Reduced hemoglobin concentration leads to anemia and tissue hypoxia.
-
Structural abnormalities cause hemoglobinopathies (e.g., sickle cell disease).
-
Altered oxygen affinity affects oxygen delivery in lung and cardiac diseases.
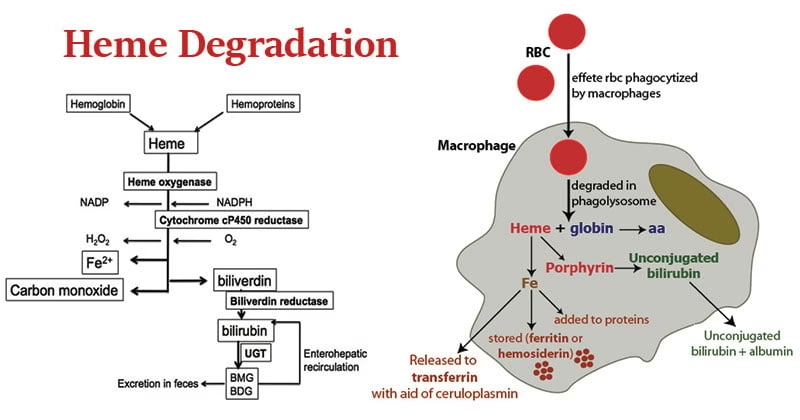
Hemoglobin Degradation
1. Lifespan and Destruction of Red Blood Cells
-
Red blood cells have an average lifespan of ~120 days.
-
Senescent or damaged RBCs lose membrane flexibility.
-
These cells are removed by macrophages of the reticuloendothelial system (RES), mainly in:
-
Spleen (primary site)
-
Liver
-
Bone marrow
-
2. Breakdown of Hemoglobin
Inside macrophages, hemoglobin is split into two major components:
A. Globin Part
-
Globin chains are hydrolyzed into amino acids.
-
Amino acids are released into circulation.
-
Reused for new protein synthesis.
B. Heme Part
Heme undergoes a series of enzymatic conversions:
Step 1: Conversion of Heme to Biliverdin
-
Enzyme: Heme oxygenase
-
Heme is opened to form biliverdin
-
Iron (Fe³⁺) is released
-
Carbon monoxide (CO) is produced as a by-product
Step 2: Conversion of Biliverdin to Bilirubin
-
Enzyme: Biliverdin reductase
-
Biliverdin is reduced to bilirubin
-
Bilirubin at this stage is unconjugated, lipid-soluble, and toxic
3. Transport of Unconjugated Bilirubin
-
Unconjugated bilirubin is transported in blood bound to albumin.
-
Albumin prevents bilirubin from crossing the blood–brain barrier.
-
Delivered to the liver for further processing.
4. Hepatic Conjugation of Bilirubin
-
In hepatocytes, bilirubin undergoes conjugation.
-
Enzyme: UDP-glucuronosyltransferase
-
Bilirubin is converted into bilirubin diglucuronide (conjugated bilirubin).
-
Conjugation makes bilirubin water-soluble and non-toxic.
5. Excretion and Intestinal Metabolism
-
Conjugated bilirubin is excreted into bile.
-
Bile enters the intestine.
-
Intestinal bacteria convert bilirubin into urobilinogen:
-
Majority → Stercobilin (excreted in feces; gives brown color)
-
Small amount → Reabsorbed → excreted as urobilin in urine (yellow color)
-
6. Iron Recycling
-
Iron released from heme is oxidized to Fe³⁺.
-
Transported in plasma by transferrin.
-
Utilized for:
-
New hemoglobin synthesis in bone marrow
-
Storage as ferritin or hemosiderin in liver and spleen
-
Table: Hemoglobin Degradation
| Component | Process | Enzyme / Carrier | End Product | Significance |
|---|---|---|---|---|
| Globin | Hydrolysis | Proteolytic enzymes | Amino acids | Reused for proteins |
| Heme → Biliverdin | Ring opening | Heme oxygenase | Biliverdin | Releases iron |
| Biliverdin → Bilirubin | Reduction | Biliverdin reductase | Unconjugated bilirubin | Transported to liver |
| Bilirubin conjugation | Glucuronidation | UDP-glucuronosyltransferase | Conjugated bilirubin | Water-soluble |
| Intestinal conversion | Bacterial action | Gut flora | Stercobilin, urobilin | Feces & urine color |
| Iron | Recycling | Transferrin | Ferritin / Hb | Prevents iron loss |
Clinical Importance of Heamoglobin
1. Role in Oxygen Delivery and Tissue Hypoxia
-
Hemoglobin determines the oxygen-carrying capacity of blood.
-
Reduced hemoglobin levels lead to tissue hypoxia, even when lung function is normal.
-
Clinical manifestations include:
-
Fatigue
-
Dyspnea
-
Palpitations
-
Poor exercise tolerance
-
-
Severe hypoxia can cause organ dysfunction, particularly in the brain and heart.
2. Anemia and Its Clinical Significance
-
Anemia is defined as a reduction in hemoglobin concentration below normal limits.
-
Causes include:
-
Iron deficiency
-
Vitamin B₁₂ or folate deficiency
-
Chronic disease
-
Bone marrow failure
-
-
Chronic anemia leads to:
-
Compensatory tachycardia
-
Cardiac hypertrophy
-
High-output heart failure (in severe cases)
-
3. Hemoglobinopathies (Structural Abnormalities)
-
Genetic defects affecting hemoglobin structure lead to hemoglobinopathies.
-
Example:
-
Sickle cell disease – abnormal β-globin structure causes RBC sickling under low oxygen tension.
-
-
Clinical effects:
-
Chronic hemolysis
-
Vaso-occlusive crises
-
Increased susceptibility to infections
-
4. Thalassemia (Disorders of Globin Chain Synthesis)
-
Caused by reduced or absent synthesis of globin chains.
-
Types:
-
α-thalassemia
-
β-thalassemia
-
-
Consequences:
-
Ineffective erythropoiesis
-
Severe anemia
-
Bone marrow expansion
-
Iron overload due to repeated transfusions
-
5. Disorders of Heme Synthesis (Porphyrias)
-
Enzyme defects in heme synthesis cause porphyrias.
-
Clinical features depend on the accumulated intermediate:
-
Abdominal pain
-
Neurological symptoms
-
Photosensitivity
-
-
Lead poisoning inhibits heme synthesis enzymes, causing:
-
Microcytic anemia
-
Neuropathy
-
Abdominal pain
-
6. Jaundice and Bilirubin Metabolism Disorders
-
Excess hemoglobin breakdown increases bilirubin production.
-
Types of jaundice:
-
Hemolytic jaundice – excess unconjugated bilirubin
-
Hepatic jaundice – impaired conjugation
-
Obstructive jaundice – blocked bile flow
-
-
Neonatal jaundice occurs due to immature liver conjugating enzymes.
7. Iron Metabolism and Storage Disorders
-
Hemoglobin degradation is the major source of body iron.
-
Disorders include:
-
Iron deficiency anemia
-
Iron overload (hemochromatosis, transfusion-related)
-
-
Abnormal iron metabolism affects:
-
Hemoglobin synthesis
-
Erythropoiesis
-
Organ function (liver, heart, pancreas)
-
8. Role in Acid–Base Balance
-
Hemoglobin is a major non-bicarbonate buffer of blood.
-
It binds hydrogen ions generated during metabolism.
-
Impaired hemoglobin buffering worsens:
-
Metabolic acidosis
-
Respiratory acidosis
-
9. Diagnostic and Laboratory Importance
-
Hemoglobin estimation is used for:
-
Screening anemia
-
Monitoring therapy response
-
Assessing nutritional status
-
-
Abnormal hemoglobin variants are detected by:
-
Electrophoresis
-
HPLC
-
Genetic testing
-
10. Cardiopulmonary and Critical Care Relevance
-
Low hemoglobin worsens outcomes in:
-
Cardiac disease
-
Respiratory failure
-
Sepsis
-
-
Hemoglobin levels guide:
-
Blood transfusion decisions
-
Oxygen therapy
-
ICU management
-
Table: Clinical Conditions Related to Hemoglobin
| Disorder | Defect | Clinical Outcome |
|---|---|---|
| Anemia | Low Hb | Tissue hypoxia |
| Thalassemia | ↓ Globin synthesis | Severe anemia |
| Sickle cell disease | Abnormal Hb structure | Hemolysis, pain |
| Porphyria | Enzyme defect | Neurocutaneous symptoms |
| Jaundice | ↑ Bilirubin | Yellow discoloration |
| Iron overload | Excess iron | Organ damage |
MCQs
1. Hemoglobin synthesis occurs mainly in:
A. Liver
B. Kidney
C. Bone marrow
D. Spleen
✅ Answer: C
2. The protein part of hemoglobin is called:
A. Heme
B. Globin
C. Porphyrin
D. Ferritin
✅ Answer: B
3. Alpha globin genes are located on chromosome:
A. 11
B. 12
C. 16
D. 18
✅ Answer: C
4. Beta globin genes are located on chromosome:
A. 16
B. 11
C. 13
D. 21
✅ Answer: B
5. Adult hemoglobin (HbA) consists of:
A. α₂γ₂
B. α₂δ₂
C. α₂β₂
D. α₄
✅ Answer: C
6. Fetal hemoglobin (HbF) consists of:
A. α₂β₂
B. α₂γ₂
C. β₄
D. α₂δ₂
✅ Answer: B
7. Globin chain synthesis occurs in:
A. Mitochondria
B. Nucleus only
C. Cytoplasm
D. Lysosomes
✅ Answer: C
8. Transcription of globin genes occurs in:
A. Cytoplasm
B. Ribosomes
C. Mitochondria
D. Nucleus
✅ Answer: D
9. Translation of globin mRNA occurs on:
A. Nucleus
B. Ribosomes
C. Golgi apparatus
D. Lysosomes
✅ Answer: B
10. Heme synthesis occurs in:
A. Cytosol only
B. Mitochondria only
C. Both mitochondria and cytosol
D. Nucleus
✅ Answer: C
11. First step of heme synthesis occurs in:
A. Cytosol
B. Mitochondria
C. Nucleus
D. Golgi
✅ Answer: B
12. Rate-limiting enzyme of heme synthesis is:
A. Ferrochelatase
B. ALA dehydratase
C. ALA synthase
D. Porphobilinogen deaminase
✅ Answer: C
13. ALA synthase requires which vitamin as cofactor?
A. Vitamin B₁
B. Vitamin B₂
C. Vitamin B₆
D. Vitamin B₁₂
✅ Answer: C
14. Substrates for ALA synthase are:
A. Glycine + Acetyl-CoA
B. Glycine + Succinyl-CoA
C. Alanine + Succinyl-CoA
D. Glycine + Propionyl-CoA
✅ Answer: B
15. ALA dehydratase is inhibited by:
A. Iron
B. Copper
C. Lead
D. Zinc
✅ Answer: C
16. Final step of heme synthesis is insertion of:
A. Zinc
B. Copper
C. Magnesium
D. Iron
✅ Answer: D
17. Enzyme inserting iron into protoporphyrin IX is:
A. ALA synthase
B. Ferrochelatase
C. Heme oxygenase
D. Biliverdin reductase
✅ Answer: B
18. Ferrochelatase is inhibited by:
A. Mercury
B. Arsenic
C. Lead
D. Cadmium
✅ Answer: C
19. Heme is synthesized mainly in:
A. Hepatocytes only
B. Erythroid cells only
C. Erythroid cells and liver
D. Kidney cells
✅ Answer: C
20. Protoporphyrin IX is formed in:
A. Cytosol
B. Mitochondria
C. Nucleus
D. Golgi
✅ Answer: B
21. Balanced synthesis of globin chains is essential to prevent:
A. Porphyria
B. Jaundice
C. Thalassemia
D. Hemochromatosis
✅ Answer: C
22. Excess unmatched globin chains cause:
A. RBC stabilization
B. Increased lifespan
C. RBC damage
D. Improved oxygen binding
✅ Answer: C
23. Globin gene switching refers to:
A. α → β chain change
B. β → γ chain change
C. γ → β chain change
D. δ → β chain change
✅ Answer: C
24. HbA₂ contains which globin chains?
A. α₂β₂
B. α₂γ₂
C. α₂δ₂
D. β₄
✅ Answer: C
25. HbA₂ percentage is increased in:
A. Iron deficiency anemia
B. β-thalassemia
C. Sickle cell disease
D. Aplastic anemia
✅ Answer: B
26. Each hemoglobin molecule contains how many heme groups?
A. One
B. Two
C. Three
D. Four
✅ Answer: D
27. Each heme group binds:
A. One oxygen molecule
B. Two oxygen molecules
C. Four oxygen molecules
D. One carbon dioxide
✅ Answer: A
28. Globin chains without heme are:
A. Stable
B. Stored
C. Rapidly degraded
D. Excreted
✅ Answer: C
29. Assembly of hemoglobin occurs in:
A. Nucleus
B. Cytoplasm
C. Golgi
D. Lysosome
✅ Answer: B
30. Normal adult hemoglobin percentage is highest for:
A. HbF
B. HbA
C. HbA₂
D. HbS
✅ Answer: B
31. Defect in globin synthesis leads to:
A. Porphyrias
B. Thalassemias
C. Jaundice
D. Hemochromatosis
✅ Answer: B
32. Defect in heme synthesis leads to:
A. Thalassemia
B. Porphyrias
C. Sickle cell disease
D. Leukemia
✅ Answer: B
33. Iron is incorporated into heme in which form?
A. Fe³⁺
B. Fe²⁺
C. Ferritin
D. Hemosiderin
✅ Answer: B
34. Which is NOT involved in hemoglobin synthesis?
A. Iron
B. Glycine
C. Succinyl-CoA
D. Albumin
✅ Answer: D
35. Heme regulates its own synthesis by inhibiting:
A. Ferrochelatase
B. ALA dehydratase
C. ALA synthase
D. Biliverdin reductase
✅ Answer: C
36. Primary site of hemoglobin synthesis in adults is:
A. Liver
B. Spleen
C. Bone marrow
D. Kidney
✅ Answer: C
37. Alpha globin chains are synthesized:
A. Only in fetal life
B. Only in adult life
C. Throughout life
D. After birth only
✅ Answer: C
38. Gamma globin chains are predominant in:
A. Adult
B. Neonatal period
C. Fetal life
D. Old age
✅ Answer: C
39. Normal hemoglobin synthesis requires all EXCEPT:
A. Iron
B. Vitamin B₆
C. Globin genes
D. Vitamin C
✅ Answer: D
40. Lead poisoning causes anemia mainly due to inhibition of:
A. Globin transcription
B. Iron absorption
C. Heme synthesis enzymes
D. RBC membrane proteins
✅ Answer: C
41. First stable porphyrin formed is:
A. Protoporphyrin IX
B. Uroporphyrinogen III
C. Coproporphyrinogen III
D. Biliverdin
✅ Answer: B
42. Heme synthesis intermediate accumulated in AIP is:
A. ALA and PBG
B. Bilirubin
C. Protoporphyrin
D. Heme
✅ Answer: A
43. Which enzyme defect causes porphyria cutanea tarda?
A. ALA synthase
B. ALA dehydratase
C. Uroporphyrinogen decarboxylase
D. Ferrochelatase
✅ Answer: C
44. Each globin chain binds heme via:
A. Covalent bond
B. Ionic bond
C. Coordination bond
D. Hydrogen bond
✅ Answer: C
45. Hemoglobin assembly requires formation of:
A. Monomers only
B. Dimers only
C. Tetramers
D. Trimers
✅ Answer: C
46. Cooperative oxygen binding is due to:
A. Heme only
B. Globin only
C. Tetrameric structure
D. Iron oxidation
✅ Answer: C
47. Site of porphobilinogen formation is:
A. Mitochondria
B. Cytosol
C. Nucleus
D. Golgi
✅ Answer: B
48. Normal hemoglobin synthesis is essential for:
A. RBC shape
B. Oxygen transport
C. CO₂ solubility
D. Plasma volume
✅ Answer: B
49. Excess iron from hemoglobin synthesis is stored as:
A. Transferrin
B. Hemosiderin
C. Ferritin
D. Bilirubin
✅ Answer: C
50. Hemoglobin synthesis defects are commonly inherited as:
A. Autosomal recessive disorders
B. Autosomal dominant disorders
C. X-linked disorders
D. Mitochondrial disorders
✅ Answer: A