
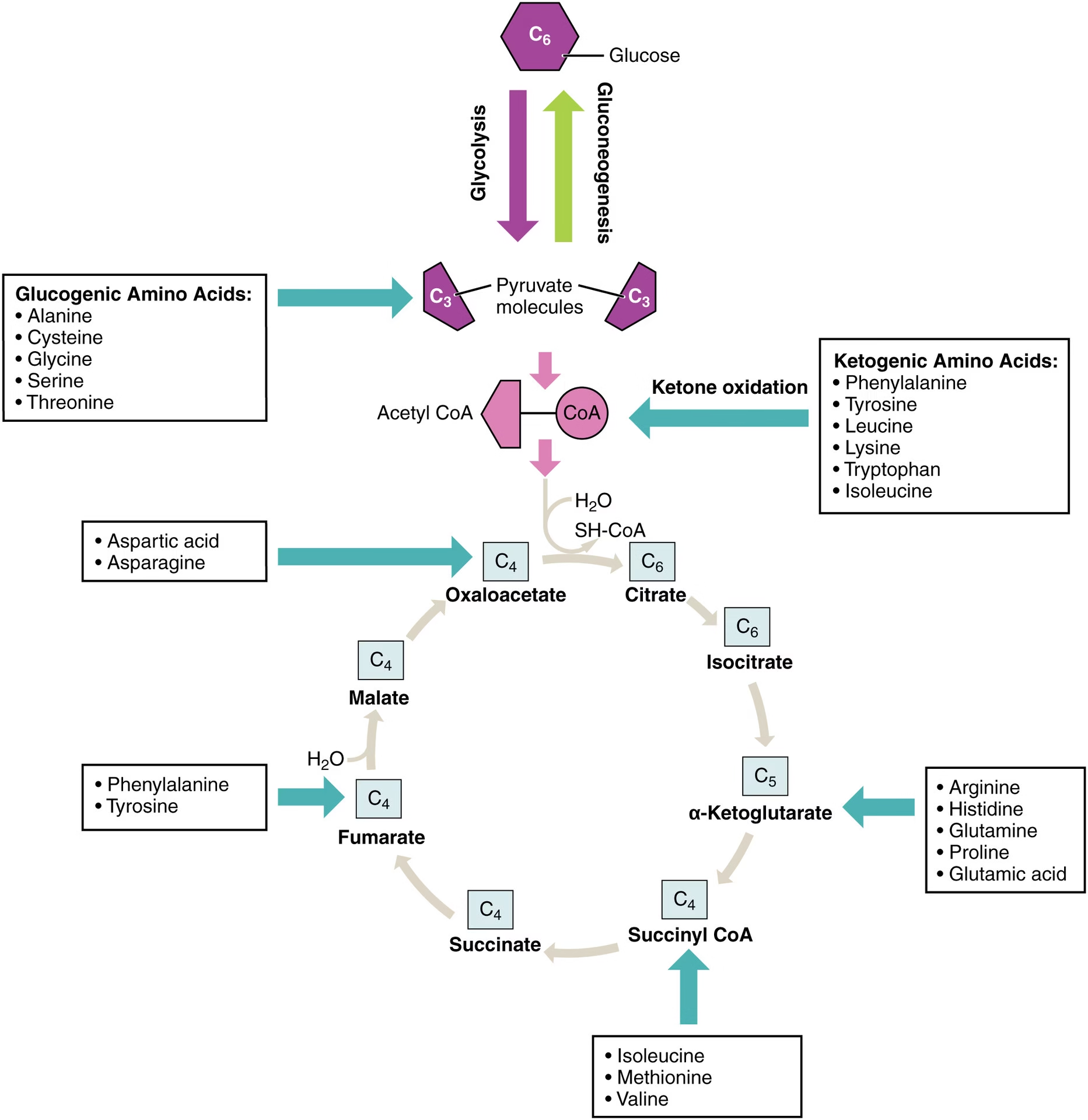
Sources of Amino Acids
-
Dietary proteins – obtained from food and digested into amino acids.
-
Degradation of body (tissue) proteins – due to normal protein turnover.
-
De novo synthesis – synthesis of non-essential amino acids from metabolic intermediates of glycolysis, TCA cycle, or pentose phosphate pathway.
-
Transamination reactions – interconversion among different amino acids.
-
Ammonia assimilation – formation of amino acids from ammonia and α-keto acids (e.g., glutamate synthesis).
Deamination:
Definition:
Deamination is the process by which an amino group (–NH₂) is removed from an amino acid, forming ammonia (NH₃) and a corresponding keto acid.
Types:
-
Oxidative Deamination: Removal of amino group with oxidation.
-
Enzyme: Glutamate dehydrogenase
-
Reaction: Glutamate + NAD⁺ + H₂O → α-Ketoglutarate + NH₃ + NADH
-
Site: Liver mitochondria
-
-
Non-oxidative Deamination: Removal of the amino group without oxidation.
-
Examples: Serine, threonine, and histidine undergo this type.
-
Importance:
-
Releases free ammonia for urea synthesis.
-
Produces keto acids for energy production, gluconeogenesis, or fatty acid synthesis.
-
Maintains nitrogen balance in the body.
Metabolism of Ammonia
1. Formation of Ammonia:
Ammonia is produced in the body from several sources:
-
Oxidative deamination of glutamate (via glutamate dehydrogenase).
-
Deamidation of glutamine and asparagine.
-
Transamination followed by deamination.
-
Bacterial action in the intestine (urease activity on urea).
2. Transport of Ammonia:
Because ammonia is toxic, it is transported in non-toxic forms:
-
As Glutamine:
-
Enzyme: Glutamine synthetase
-
NH₃ + Glutamate → Glutamine (in peripheral tissues)
-
Glutamine travels to the liver or kidney where it is hydrolyzed back to NH₃.
-
-
As Alanine:
-
Formed in muscle via alanine transaminase (ALT).
-
Alanine carries ammonia to the liver for conversion to urea (Glucose–Alanine cycle).
-
3. Detoxification and Excretion:
-
In Liver:
-
Ammonia is converted into urea by the urea cycle (main pathway of detoxification).
-
-
In Kidney:
-
Small amount of ammonia is directly excreted in urine as NH₄⁺ to help maintain acid–base balance.
-
4. Utilization:
-
Small amounts are used in the synthesis of amino acids, purines, pyrimidines, and glutamine.
5. Toxicity:
-
Excess ammonia causes hyperammonemia, leading to CNS symptoms (confusion, tremor, coma).
-
Normally, blood ammonia is kept below 50 µmol/L by efficient liver function.
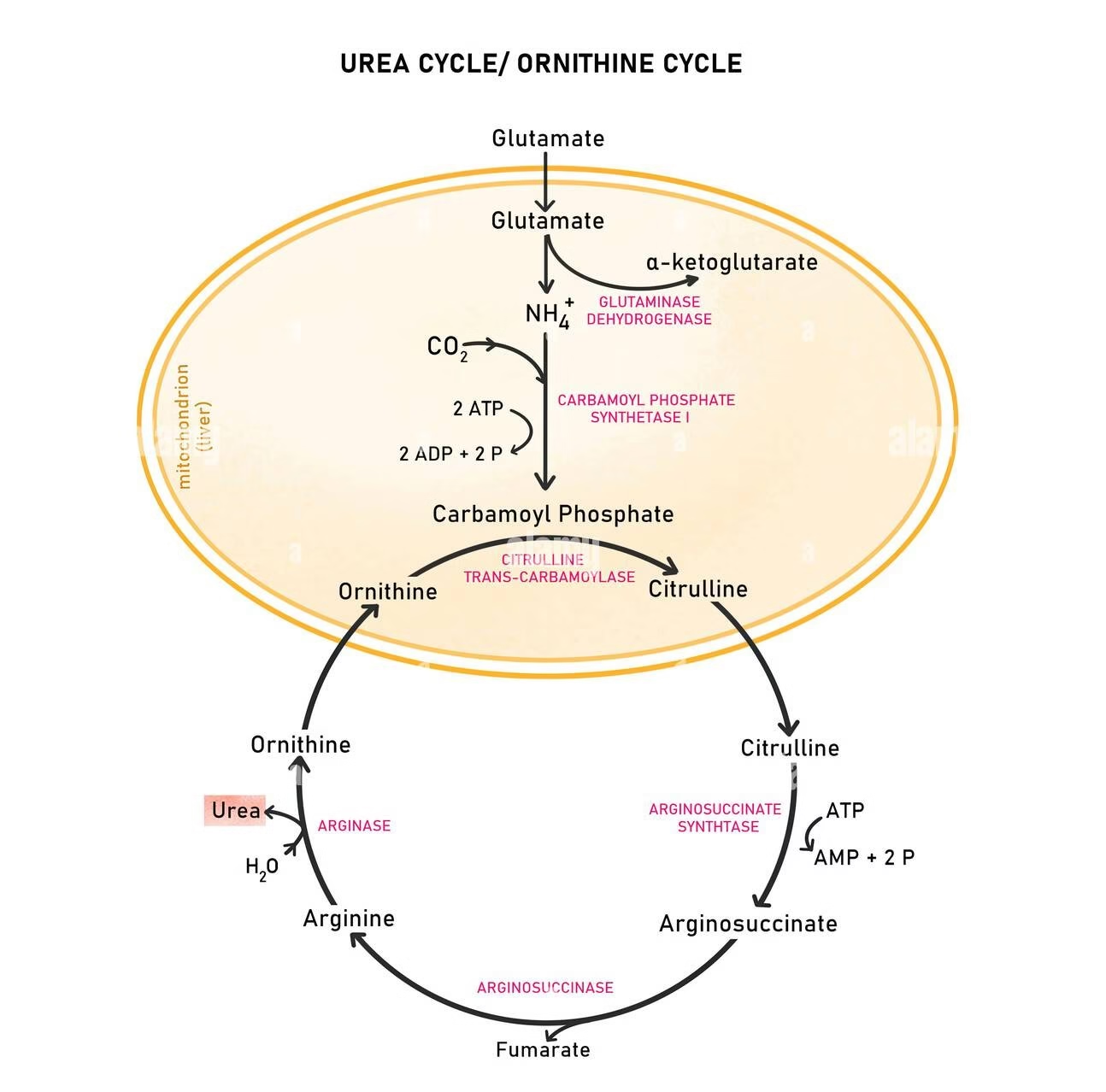
Formation of Urea
Definition:
- The urea cycle (also called the ornithine cycle) is the biochemical pathway in the liver by which toxic ammonia (NH₃), produced from amino acid catabolism, is converted into non-toxic urea, which is then excreted by the kidneys.
- It was discovered by Hans Krebs and Kurt Henseleit in 1932 and represents the first cyclic metabolic pathway identified.
Site of Occurrence:
-
Organ: Liver (hepatocytes)
-
Subcellular Location:
-
Mitochondria: First two reactions
-
Cytosol: Last three reactions
-
Precursors of Urea:
Urea contains two nitrogen atoms and one carbon atom:
| Atom | Source |
|---|---|
| One nitrogen | From ammonia (NH₃) produced by oxidative deamination of glutamate |
| Second nitrogen | From aspartate formed by transamination of oxaloacetate |
| Carbon atom | From CO₂ (as bicarbonate, HCO₃⁻) |
Steps of Urea Formation:
Step 1 – Formation of Carbamoyl Phosphate
-
Enzyme: Carbamoyl phosphate synthetase I (CPS I)
-
Location: Mitochondria
-
Reaction:
NH3+CO2+2ATP→Carbamoyl phosphate+2ADP+Pi
-
Cofactor: N-Acetylglutamate (NAG) – an essential allosteric activator.
-
Significance: This is the rate-limiting step of the cycle.
Step 2 – Formation of Citrulline
-
Enzyme: Ornithine transcarbamoylase (OTC)
-
Location: Mitochondria
-
Reaction:
Ornithine+Carbamoyl phosphate→Citrulline+Pi
-
Process: Citrulline is then transported to the cytosol via an ornithine–citrulline antiporter.
Step 3 – Formation of Argininosuccinate
-
Enzyme: Argininosuccinate synthetase
-
Location: Cytosol
-
Reaction:
Citrulline+Aspartate+ATP→Argininosuccinate+AMP+PPi
-
Significance:
-
Aspartate contributes the second nitrogen of urea.
-
Uses two high-energy phosphate bonds (ATP → AMP).
-
Step 4 – Cleavage of Argininosuccinate
-
Enzyme: Argininosuccinate lyase (Argininosuccinase)
-
Location: Cytosol
-
Reaction:
Argininosuccinate→Arginine+Fumarate
-
Significance:
-
Fumarate enters the TCA cycle, forming malate and oxaloacetate (link between the two cycles).
-
This connection is known as the Aspartate–Argininosuccinate shunt.
-
Step 5 – Formation of Urea and Regeneration of Ornithine
-
Enzyme: Arginase
-
Location: Cytosol
-
Reaction:
Arginine+H2O→Urea+Ornithine
-
Significance:
-
Urea is released into blood → transported to kidneys → excreted in urine.
-
Ornithine is recycled back into mitochondria to continue the cycle.
-
Overall Reaction:
2NH3+CO2+3ATP+H2O→Urea+2ADP+AMP+4Pi+
-
Energy cost: 3 ATP (4 high-energy bonds) are used for each molecule of urea synthesized.
-
Energy recovery: Oxidation of fumarate via TCA cycle yields ~1 NADH (≈ 3 ATP), partly compensating energy expenditure.
Regulation of Urea Cycle:
-
Allosteric Regulation:
-
CPS I is activated by N-Acetylglutamate (NAG).
-
Arginine stimulates NAG synthesis → increases urea cycle activity.
-
-
Substrate Availability:
-
Increased ammonia or amino acid load enhances urea formation.
-
-
Enzyme Induction:
-
High-protein diet or fasting induces the synthesis of urea cycle enzymes.
-
Physiological Significance:
-
Ammonia detoxification: Converts toxic NH₃ to non-toxic urea.
-
Nitrogen excretion: Main pathway for nitrogen elimination.
-
Interconnection with energy metabolism: Fumarate links to the TCA cycle.
-
Maintenance of acid–base balance: Prevents ammonia accumulation, which raises pH.
Clinical Correlations:
1. Hyperammonemia:
-
Elevated blood ammonia due to defective urea formation.
-
Causes:
-
Liver disease (acquired)
-
Congenital enzyme deficiencies (inherited)
-
2. Enzyme Deficiencies and Disorders:
| Enzyme Deficiency | Disorder | Key Features |
|---|---|---|
| CPS I | Hyperammonemia Type I | ↑ NH₃, ↓ citrulline |
| OTC | Hyperammonemia Type II | ↑ NH₃, ↑ orotic acid (X-linked) |
| Argininosuccinate synthetase | Citrullinemia | ↑ Citrulline |
| Argininosuccinate lyase | Argininosuccinic aciduria | ↑ Argininosuccinate |
| Arginase | Hyperargininemia | ↑ Arginine, neurological symptoms |
3. Symptoms:
Vomiting, lethargy, seizures, cerebral edema, coma.
4. Treatment:
-
Dietary: Low-protein diet
-
Drugs: Sodium benzoate, phenylacetate, or phenylbutyrate (bind excess ammonia)
-
Supplement: Arginine or citrulline (depending on deficiency)
-
Severe cases: Liver transplantation
Quantitative Aspect:
-
Daily urea excretion: 25–30 g/day in adults.
-
Constitutes about 80–90% of total urinary nitrogen.
Link with Other Metabolic Pathways:
| Cycle/Pathway | Connection |
|---|---|
| TCA Cycle | Fumarate from urea cycle enters TCA; CO₂ from TCA used in CPS I reaction. |
| Amino Acid Metabolism | Provides ammonia and aspartate. |
| Transamination Reactions | Form aspartate and glutamate, key intermediates. |
Mnemonic for Enzymes (in order):
C – O – A – A – A
-
-
C – Carbamoyl phosphate synthetase I
-
O – Ornithine transcarbamoylase
-
A – Argininosuccinate synthetase
-
A – Argininosuccinate lyase
-
A – Arginase
-
Glycine Metabolism
Introduction
-
Glycine is the simplest amino acid (NH₂-CH₂-COOH).
-
It is non-essential, glucogenic, and plays key roles in protein synthesis, one-carbon metabolism, and biosynthesis of important biomolecules.
| Feature | Description |
|---|---|
| Chemical Formula | C₂H₅NO₂ |
| Molecular Weight | 75 Da |
| Nature | Non-essential, glucogenic amino acid |
| Chirality | Achiral (only amino acid without optical activity) |
| Side Chain | Hydrogen (–H) |
| Solubility | Highly water-soluble |
| Special Feature | Found abundantly in collagen (every 3rd residue) |
Sources / Synthesis of Glycine
Glycine can be synthesized endogenously in several ways:
| Pathway | Enzyme | Cofactors | Location | Significance |
|---|---|---|---|---|
| Serine → Glycine | SHMT | PLP, THF | Cytosol & mitochondria | Major contributor; linked to folate cycle |
| Threonine → Glycine | Threonine aldolase | PLP | Cytosol | Minor physiological source |
| Choline → Betaine → Glycine | Dimethylglycine dehydrogenase | FAD | Mitochondria | Connects methylation cycle |
| Glyoxylate → Glycine | AGT | PLP | Peroxisomes | Defects → Primary hyperoxaluria |
Cofactors:
-
Pyridoxal phosphate (Vitamin B₆)
-
Tetrahydrofolate (THF) for one-carbon transfer
Catabolism of Glycine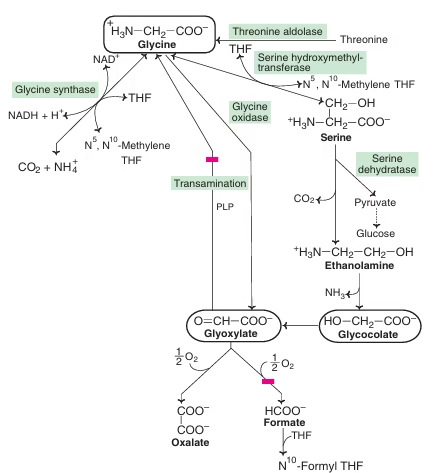
Metabolic Roles of Glycine
| Function | Compound Synthesized | Enzyme (if applicable) |
|---|---|---|
| Heme synthesis | Glycine + Succinyl-CoA → δ-Aminolevulinic acid (ALA) | ALA synthase |
| Creatine synthesis | Glycine + Arginine → Guanidinoacetate → Creatine | Transamidase |
| Purine synthesis | Donates C₄, C₅, N₇ atoms of purine ring | — |
| Glutathione synthesis | Glycine + Cysteine + Glutamate → GSH | Glutathione synthetase |
| Bile salt conjugation | Bile acids + Glycine → Glycocholic acid, etc. | Bile acid–CoA:amino acid N-acyltransferase |
| Porphyrin & Heme | As above | — |
| One-carbon metabolism | Forms CH₂-THF via glycine cleavage system | — |
Regulation of Glycine Metabolism
| Regulator | Effect on Metabolism | Notes |
|---|---|---|
| PLP (Vitamin B6) | Required for SHMT, transaminases, ALA synthase | Deficiency ↓ glycine processing |
| Folate (THF) | Necessary for serine–glycine conversion | Folate deficiency disrupts 1-carbon metabolism |
| GCS Activity | Controls glycine degradation | Deficiency → NKH |
| Dietary Protein | ↑ glycine levels | Protein-rich foods boost supply |
| Hormones | Glucagon ↑ catabolism, Insulin ↑ anabolism | Affects amino acid turnover |
| Peroxisomal enzymes | Regulate glyoxylate handling | Defects → Hyperoxaluria |
Clinical Significance
| Disorder | Enzyme Defect / Cause | Key Features |
|---|---|---|
| Non-ketotic hyperglycinemia (glycine encephalopathy) | Defect in glycine cleavage enzyme complex | ↑ Glycine in CSF & plasma → severe neurological symptoms, seizures, mental retardation |
| Primary hyperoxaluria | Defective glyoxylate metabolism (↑ glyoxylate → oxalate) | Kidney stones, renal failure |
| Deficiency of THF or B₆ | Impaired glycine metabolism | Anemia, reduced one-carbon transfer reactions |
Laboratory Diagnosis
| Test | Sample | Purpose |
|---|---|---|
| Plasma amino acid analysis | Blood | Detect elevated glycine |
| CSF amino acid profiling | CSF | Diagnose NKH |
| Urine oxalate measurement | Urine | Diagnose hyperoxaluria |
| Hippurate measurement | Urine | Evaluate detoxification function |
| HPLC / GC-MS | Plasma/Urine | Accurate quantitative analysis |
Metabolism of Phenylalanine and Tyrosine
Introduction
-
Phenylalanine (Phe) and Tyrosine (Tyr) are aromatic amino acids derived from the shikimate pathway in plants and obtained in humans from diet.
-
Phenylalanine is an essential amino acid, while tyrosine is non-essential (formed from phenylalanine).
-
Both are glucogenic and ketogenic.
-
They serve as precursors for several vital molecules — catecholamines (dopamine, norepinephrine, epinephrine), thyroid hormones, and melanin.
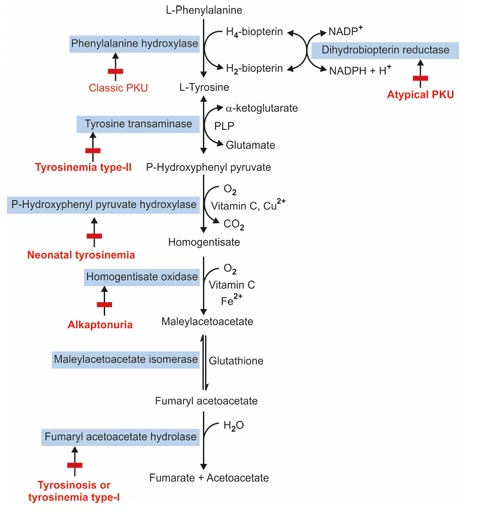
Conversion of Phenylalanine to Tyrosine:
| Component | Description |
|---|---|
| Enzyme | Phenylalanine hydroxylase (PAH) |
| Location | Liver |
| Cofactor | BH₄, Fe²⁺ |
| Importance | Prevents toxic buildup of phenylalanine |
| Defects | Cause Phenylketonuria (PKU) |
Reaction:Phenylalanine+O2+Tetrahydrobiopterin(BH4)→Tyrosine+H2O+Dihydrobiopterin(BH2)
Enzyme: Phenylalanine hydroxylase
Cofactors:
- Tetrahydrobiopterin (BH₄) – acts as a reducing cofactor
- Fe²⁺ (Iron) – required for enzyme activity
- Oxygen (O₂) – provides one atom for hydroxylation
Location: Liver cytosol
Mechanism:
- The enzyme adds a hydroxyl group (–OH) to the para position of the benzene ring of phenylalanine.
- This converts phenylalanine into tyrosine, making it hydroxylated at the 4th position (p-hydroxyphenylalanine).
- During the reaction, BH₄ is oxidized to BH₂ and later regenerated by dihydropteridine reductase using NADPH.
Significance:
- This is the first step in phenylalanine catabolism.
- It converts the essential amino acid (phenylalanine) into a non-essential amino acid (tyrosine).
- Tyrosine then serves as a precursor for melanin, catecholamines (dopamine, epinephrine, norepinephrine), and thyroid hormones.
Clinical Importance:
- Deficiency of phenylalanine hydroxylase or BH₄ causes Phenylketonuria (PKU) → accumulation of phenylalanine and its toxic metabolites leading to mental retardation and hypopigmentation.
Regulation of Phenylalanine and Tyrosine Metabolism
| Regulation Type | Molecules Involved | Effect |
|---|---|---|
| Feedback inhibition | High tyrosine | Inhibits phenylalanine hydroxylase |
| Hormonal | Glucocorticoids | Induce tyrosine aminotransferase |
| Cofactor availability | BH₄, PLP, Vit C | Controls specific enzymes |
| Genetic | PAH, HGD, FAH mutations | Cause metabolic disorders |
Metabolic Disorders of Phenylalanine and Tyrosine
Phenylketonuria (PKU) is an inborn error of phenylalanine metabolism associated with the inability to convert phenylalanine to tyrosine. Ratio 1 in 20,000 newborns.
| Types | Condition | Enzyme defects |
| Type 1 | Classical type of PKU | Phenylalanine hydroxylase enzyme deficiency |
| Type 2 | Persistent hyperphenylalaninaemia | Phenylalanine hydroxylase enzyme deficiency |
| Type 3 | Transient mild hyperphenylalaninaemia | Phenylalanine hydroxylase enzyme is delayed |
| Type 4 | Dihydropterine reductase deficiency | Dihydropterine deficiency |
| Type 5 | Abnormal Dihydropterine function | Dihydropterine synthesis defects |
Tyrosinemia
There are three types of tyrosinemia:
- Tyrosinemia type-I (Tyrosinosis/Hepatorenal tyrosinemia)
- Tyrosinemia type-II (Richner-Hanhart syndrome)
- Neonatal tyrosinemia
Alkaptonuria
Definition: Alkaptonuria is a rare autosomal recessive metabolic disorder characterized by the accumulation of homogentisic acid due to a deficiency in the enzyme homogentisate oxidase.
Enzyme Defect
- The defective enzyme in alkaptonuria is homogentisate oxidase in tyrosine metabolism.
- Homogentisate accumulates in tissues and blood and is excreted into urine. The urine of alkaptonuria patients resembles coke in colour.
Biochemical Manifestations
- Homogentisic Acid Accumulation:
- The main biochemical defect is the elevated levels of homogentisic acid in the body.
- This compound is toxic and can lead to various pathological effects.
- Urine Color Change:
- Urine from affected individuals darkens upon exposure to air due to the oxidation of homogentisic acid. This can happen within a few hours and is a hallmark feature of the disease.
- Freshly voided urine may appear normal but darkens rapidly when left standing.
- Ochronosis:
- Chronic accumulation of homogentisic acid can lead to tissue deposits in connective tissues, known as ochronosis.
- Common sites include the cartilage of joints, intervertebral discs, and the skin. This can cause discolouration and degenerative joint disease.
- Systemic Effects:
- Patients may experience early-onset arthritis, especially in large joints (e.g., hips, knees).
- Other complications include potential heart valve issues and kidney stones.
Diagnosis
- Clinical Evaluation:
- Diagnosis often starts with a clinical suspicion based on symptoms such as dark urine and joint pain.
- Urine Analysis:
- Colour Test: The darkening of urine upon standing is a critical diagnostic sign.
- Chemical Analysis: Urine can be tested for the presence of homogentisic acid using qualitative and quantitative methods, such as:
- HPLC (High-Performance Liquid Chromatography): Measures the levels of homogentisic acid.
- Spot Tests: A simple qualitative test where a few drops of urine can react with specific reagents to indicate the presence of homogentisic acid.
- Genetic Testing:
- Identification of mutations in the HGD gene can confirm the diagnosis.
- Genetic counselling may be recommended for affected individuals and their families.
Management
While there is currently no cure for alkaptonuria, management focuses on symptomatic relief and preventing complications:
- Lifestyle Modifications:
- Encourage a balanced diet with limited intake of phenylalanine and tyrosine, although dietary restrictions may vary in severity based on individual cases.
- Maintain hydration to help reduce the risk of kidney stones.
- Pain Management:
- Nonsteroidal anti-inflammatory drugs (NSAIDs) can be used to manage joint pain.
- In severe cases, physical therapy or joint replacement surgery may be necessary.
- Monitoring:
- Regular followup to monitor joint health and function.
- Periodic assessment of urine for homogentisic acid levels can help gauge the condition’s progression.
- Research and Experimental Therapies:
- Ongoing research explores potential treatments, including enzyme replacement therapy and dietary supplements, but these are not yet standard practice.
Phenylketonuria
Phenylketonuria (PKU) is a genetic metabolic disorder caused by a defect in the enzyme phenylalanine hydroxylase (PAH). This condition affects the body’s ability to metabolize the amino acid phenylalanine, leading to various biochemical and clinical manifestations.
Enzyme Defect
- Enzyme: Phenylalanine hydroxylase (PAH)
- Function: PAH catalyzes the conversion of phenylalanine to tyrosine, another amino acid.
- Deficiency: When PAH is deficient or absent, phenylalanine accumulates in the body, leading to toxic effects, particularly in the brain. The incidence of PKU is 1 in 10,000 births.
Biochemical Manifestations
- Elevated Phenylalanine Levels:
- The hallmark of PKU is significantly increased levels of phenylalanine in the blood (hyperphenylalaninemia).
- Normal phenylalanine levels are usually between 0.5 to 1.5 mg/dL, while levels in untreated PKU can exceed 20 mg/dL.
- Deficiency of Tyrosine:
- Since PAH converts phenylalanine to tyrosine, its deficiency leads to reduced levels of tyrosine, which is essential for neurotransmitter synthesis (dopamine, norepinephrine).
- Metabolite Accumulation:
- Increased phenylalanine can be converted to phenylpyruvate, which is then excreted in urine, along with other phenylalanine derivatives.
- Neurological Effects:
- High levels of phenylalanine are neurotoxic, leading to developmental delays, intellectual disability, seizures, and behavioural problems if untreated.
- Other Symptoms:
- Patients may develop lighter skin and hair due to reduced melanin production (tyrosine is a precursor for melanin).
Diagnosis
- Newborn screening:
- PKU is typically diagnosed through routine newborn screening programs that measure blood phenylalanine levels.
- A heel prick test is performed shortly after birth, usually within the first week.
- Blood Tests:
- Phenylalanine Levels: A blood sample is analyzed for elevated levels of phenylalanine. A level above the threshold indicates a risk for PKU.
- Tandem Mass Spectrometry: This advanced technique can confirm elevated phenylalanine and is often used in newborn screening.
- Genetic Testing:
- Confirmatory testing can involve genetic analysis to identify mutations in the PAH gene, confirming the diagnosis and subtype of PKU.
- This testing can also help assess the risk for family members.
- Clinical Evaluation:
- If PKU is suspected, a thorough clinical assessment will be conducted, looking for signs of neurological impairment or developmental delays.
Management
- Dietary Management:
- The cornerstone of PKU management is a strict, lifelong low-phenylalanine diet.
- Patients avoid high-protein foods (meat, fish, eggs, dairy, nuts) and certain grains.
- Special medical formulas that provide essential nutrients without phenylalanine are often used.
- Monitoring:
- Monitoring blood phenylalanine levels is crucial to ensure they remain within target ranges, typically below 6 mg/dL.
- Dietary adjustments may be necessary based on these levels.
- Supplementation:
- Tyrosine supplementation may be necessary due to its reduced levels in PKU patients.
- Emerging Therapies:
- New treatments, such as enzyme replacement therapy, pharmacological therapies (e.g., sapropterin dihydrochloride), and gene therapy, are being researched and may offer additional options.
- Support Services:
- Nutritional counselling and support groups can provide essential education and emotional support for families managing the condition.
Maple Syrup Urine Disease
Maple Syrup Urine Disease (MSUD) is a rare genetic metabolic disorder caused by a defect in the branched-chain alpha-keto acid dehydrogenase (BCKAD) complex, which is essential for the metabolism of branched-chain amino acids (BCAAs): leucine, isoleucine, and valine.
Enzyme Defect
- Enzyme Complex: Branched-chain alpha-keto acid dehydrogenase (BCKAD) Complex
- Gene Mutations: Mutations can occur in several genes that encode components of the BCKAD complex, including:
- BCKDHA (alpha component)
- BCKDHB (beta component)
- DBT (dihydrolipoamide branched-chain transacylase)
- Function: The BCKAD complex catalyzes the oxidative decarboxylation of branched-chain alpha-keto acids derived from the BCAAs.
- Deficiency: When this complex is deficient, branched-chain amino acids accumulate and their corresponding alpha-keto acids in the blood and urine.
Biochemical Manifestations
- Elevated Branched-Chain Amino Acids:
- Blood levels of leucine, isoleucine, and valine become significantly elevated. Normal levels are typically below 150 μmol/L for leucine, 40 μmol/L for isoleucine, and 100 μmol/L for valine.
- In untreated MSUD, leucine levels can exceed 1,000 μmol/L.
- Accumulation of Alpha-Keto Acids:
- Alongside elevated BCAAs, their corresponding alpha-keto acids (such as alpha-ketoisocaproic acid) accumulate, which can be toxic, especially to the nervous system.
- Neurological Symptoms:
- Toxic levels of BCAAs and their metabolites can lead to neurological issues, including lethargy, seizures, and developmental delays.
- Urine Characteristics:
- The condition is named for the sweet, maple syrup-like odour of the urine due to the presence of branched-chain keto acids.
Diagnosis
- Newborn Screening:
- MSUD is typically diagnosed through routine newborn screening programs, which test for elevated levels of leucine and other BCAAs in dried blood spots collected shortly after birth.
- Clinical Presentation:
- Symptoms often appear within the first few days of life, including poor feeding, vomiting, lethargy, and irritability.
- Blood Tests:
- Confirmatory blood tests measure the levels of branched-chain amino acids, showing significant elevations of leucine, isoleucine, and valine.
- Urine Analysis:
- Urinalysis may reveal the presence of branched-chain keto acids, which specific chemical tests can detect.
- Genetic Testing:
- Genetic testing can confirm the diagnosis by identifying mutations in the genes associated with the BCKAD complex.
- This testing can also help determine the specific subtype of MSUD, as there are several variants (classic, intermediate, and thiamine-responsive).
Management
- Dietary Management:
- The primary treatment for MSUD involves a strict diet low in branched-chain amino acids, particularly leucine.
- Special medical formulas that provide essential amino acids without BCAAs are essential for growth and development.
- Monitoring:
- Regularly monitoring blood amino acid levels is crucial to prevent toxic accumulation and adjust dietary intake as needed.
- Emergency Protocols:
- In times of illness or stress, rapid intervention may be required to manage acute metabolic crises, which can be life-threatening. This often involves hospitalization and intravenous fluids.
- Potential Therapies:
- Research is ongoing into new treatments, including enzyme replacement therapy, gene therapy, and alternative dietary strategies.
- Support Services:
- Nutritional counselling and family support resources are vital for managing the condition effectively.
Albinism
Albinism is a group of genetic disorders characterized by a deficiency or absence of melanin production in the skin, hair, and eyes. The condition arises from defects in specific enzymes involved in the melanin biosynthesis pathway.
Enzyme Defect
- Common Enzyme Defects:
- Tyrosinase: The most common defect occurs in tyrosinase, which catalyzes the conversion of tyrosine to DOPA (dihydroxyphenylalanine) and then to dopaquinone, a melanin precursor. This is associated with Oculocutaneous Albinism Type 1 (OCA1).
- Other Enzymes: Defects in other enzymes like tyrosinase-related protein 1 (TYRP1) and Dopachrome tautomerase (DCT) lead to other forms of albinism.
- Gene Mutations:
- TYR (tyrosinase gene), OCA2 (associated with OCA2), and TYRP1 genes are among the most frequently mutated genes in different types of albinism.
Biochemical Manifestations
- Reduced Melanin Production:
- Affected individuals have significantly reduced or absent melanin levels in the skin, hair, and eyes.
- The lack of melanin leads to lighter pigmentation and can result in white or light-coloured hair and skin.
- Ocular Abnormalities:
Common ocular manifestations include:
-
- Nystagmus: Involuntary eye movements.
- Strabismus: Misalignment of the eyes.
- Photophobia: Sensitivity to bright light.
- Reduced Visual Acuity: Impaired vision due to improper retina development.
3. Increased Sun Sensitivity:
-
- Individuals with albinism are more susceptible to sunburn and skin damage due to the lack of protective melanin.
- They have a higher risk of developing skin cancers, including melanoma.
Diagnosis
- Clinical Evaluation:
- Diagnosis often begins with a clinical examination that reveals characteristic features such as light skin, hair, eye colour, and ocular abnormalities.
- Family History:
- A family history of albinism can support the diagnosis, as many forms are inherited in an autosomal recessive manner.
- Genetic Testing:
- Molecular genetic testing can confirm the diagnosis by identifying mutations in the relevant genes.
- This testing can also help determine the specific type of albinism.
- Ophthalmologic Examination:
- A detailed eye examination can reveal specific ocular defects associated with albinism, such as foveal hypoplasia (underdevelopment of the fovea) and abnormal retinal structure.
- Skin Biopsy:
- Sometimes, a skin biopsy may be performed to assess melanin production and distribution.
Management
- Sun Protection:
- Individuals with albinism should take strict measures to protect their skin from UV exposure, including high-SPF sunscreen, protective clothing, and sunglasses.
- Vision Support:
- Visual aids and corrective lenses may be necessary to improve visual acuity.
- Regular eye exams are essential for monitoring and addressing ocular issues.
- Educational Support:
- Specialized educational resources may be required to accommodate visual impairments.
- Psychosocial Support:
- Counselling and support groups can help individuals and families cope with the challenges associated with living with albinism, including social stigma and psychological impacts.
Hartnup disorder
Hartnup disorder is a rare genetic condition characterized by the impaired absorption of certain amino acids, primarily neutral ones, in the kidneys and intestines. A defect in a specific transporter protein causes this disorder.
Enzyme Defect
- Transporter Defect: The primary defect in Hartnup disorder is in the gene, which encodes a sodium-dependent neutral amino acid transporter.
- Affected Transport: This transporter reabsorbs neutral amino acids (such as tryptophan, leucine, isoleucine, and phenylalanine) in the renal tubules and the intestines.
Biochemical Manifestations
- Amino Aciduria:
- Due to the defective transporter, neutral amino acids are not effectively reabsorbed, leading to excessive urine loss (aminoaciduria).
- This results in low blood levels of these amino acids (hypoaminoacidemia).
- Tryptophan Deficiency:
- The loss of tryptophan can lead to decreased serotonin and niacin (vitamin B3) synthesis, as tryptophan is a precursor for both.
- Niacin deficiency can result in symptoms similar to pellagra, including diarrhoea, dermatitis, and dementia.
- Neurological Symptoms:
- Some individuals may experience neurological issues due to low levels of neurotransmitters (e.g., serotonin) derived from tryptophan. Symptoms can include ataxia, psychiatric disturbances, and mood changes.
- Skin Manifestations:
- Some patients may develop photosensitivity and skin rashes, especially when exposed to sunlight, due to the effects of tryptophan deficiency and niacin deficiency.
Diagnosis
- Clinical Evaluation:
- Diagnosis often begins with a clinical assessment of photosensitivity, ataxia, and neurological manifestations.
- Family history may provide additional context, as Hartnup disorder is inherited in an autosomal recessive manner.
- Urine Analysis:
- A 24-hour urine collection can reveal elevated levels of neutral amino acids, particularly tryptophan, leucine, and isoleucine.
- Blood Tests:
- Blood tests may show low levels of neutral amino acids, especially tryptophan.
- Genetic Testing:
- Molecular genetic testing can confirm the diagnosis by identifying mutations in the SLC6A19
- Genetic counselling may be recommended for affected individuals and their families.
- Response to Niacin Supplementation:
- Sometimes, a trial of niacin supplementation can help assess the impact of the deficiency on symptoms and provide supportive evidence for the diagnosis.
Management
- Dietary Management:
- A balanced diet rich in proteins and potentially supplemented with essential amino acids may help manage symptoms.
- Some individuals may benefit from a niacin-rich diet or supplementation to prevent deficiency.
- Symptomatic Treatment:
- Addressing specific symptoms, such as skin rashes or neurological issues, may require additional treatment and support.
- Sun Protection:
- Patients may need to avoid excessive sun exposure and use sunscreen to manage photosensitivity.
- Regular Monitoring:
- Regular follow-ups with healthcare providers are important to monitor amino acid levels and overall health.
MCQs
1. The first step in dietary protein digestion begins in the:
A. Mouth
B. Stomach
C. Duodenum
D. Ileum
2. The major proteolytic enzyme of the stomach is:
A. Trypsin
B. Pepsin
C. Chymotrypsin
D. Elastase
3. Pepsinogen is activated to pepsin by:
A. Trypsin
B. HCl
C. Secretin
D. Bicarbonate
4. Enteropeptidase converts:
A. Trypsin to trypsinogen
B. Trypsinogen to trypsin
C. Pepsinogen to pepsin
D. Proelastase to elastase
5. The major site of amino acid absorption is:
A. Stomach
B. Duodenum
C. Jejunum
D. Colon
6. Amino acids are absorbed by:
A. Primary active transport
B. Secondary active transport
C. Diffusion
D. Facilitated diffusion
7. Transamination requires which coenzyme?
A. NAD⁺
B. PLP (Vitamin B6)
C. Biotin
D. THF
8. The major enzyme for removing the amino group from glutamate is:
A. ALT
B. AST
C. Glutamate dehydrogenase
D. Transaminase
9. Glutamate dehydrogenase uses which cofactors?
A. NAD⁺ or NADP⁺
B. FAD
C. THF
D. PLP
10. Urea cycle occurs primarily in the:
A. Brain
B. Kidney
C. Liver
D. Intestine
11. The first amino acid used in urea cycle is:
A. Glycine
B. Glutamine
C. Arginine
D. Ammonia
12. Carbamoyl phosphate synthase I is located in:
A. Cytosol
B. Mitochondria
C. ER
D. Nucleus
13. The rate-limiting enzyme of urea cycle is:
A. Arginase
B. CPS-I
C. ASS
D. ASL
14. CPS-I requires which activator?
A. Glutamate
B. Aspartate
C. N-Acetylglutamate
D. Fumarate
15. Ornithine transcarbamylase (OTC) deficiency leads to:
A. Hyperglycinemia
B. Hyperammonemia
C. Maple syrup urine disease
D. Hartnup disease
16. In the liver, ammonia is converted to:
A. Uric acid
B. Creatinine
C. Urea
D. Glucose
17. During prolonged fasting, major gluconeogenic amino acid is:
A. Phenylalanine
B. Leucine
C. Alanine
D. Tryptophan
18. Glucogenic amino acids produce:
A. Acetoacetate
B. Acetyl-CoA
C. TCA cycle intermediates
D. Fatty acids
19. Ketogenic amino acids include:
A. Leucine & Lysine
B. Alanine & Glycine
C. Valine & Proline
D. Histidine & Arginine
20. Which amino acid forms serotonin?
A. Tyrosine
B. Tryptophan
C. Phenylalanine
D. Histidine
21. Phenylalanine hydroxylase requires:
A. Biotin
B. THF
C. BH4
D. PLP
22. Transamination of alanine produces:
A. Acetyl-CoA
B. Pyruvate
C. Oxaloacetate
D. α-Ketoglutarate
23. Glutamine serves as a major carrier of:
A. Hydrogen ions
B. CO₂
C. Ammonia
D. Uric acid
24. Cystinuria is caused by defective transport of:
A. Neutral amino acids
B. Acidic amino acids
C. Basic amino acids & cystine
D. Aromatic amino acids
25. Maple syrup urine disease involves defect in metabolism of:
A. Aromatic amino acids
B. Sulfur-containing amino acids
C. Branched-chain amino acids
D. Acidic amino acids
26. Alkaptonuria is due to defect in:
A. Phenylalanine hydroxylase
B. Homogentisate oxidase
C. Tyrosinase
D. DOPA decarboxylase
27. Phenylketonuria results from deficiency of:
A. BH2
B. CPS-I
C. Phenylalanine hydroxylase
D. Tryptophan hydroxylase
28. Carbamoyl phosphate is formed from:
A. CO₂ + NH₃ + ATP
B. CO₂ + H₂O + ATP
C. Glutamine + ATP
D. Urea + ATP
29. The step in urea cycle that releases urea is catalyzed by:
A. ASS
B. ASL
C. CPS-I
D. Arginase
30. Fumarate formed in urea cycle enters:
A. Glycolysis
B. TCA cycle
C. PPP
D. FA synthesis
31. Nitrogen balance is positive in:
A. Illness
B. Fasting
C. Growth & pregnancy
D. Burns
32. Kwashiorkor is characterized by:
A. Edema
B. Muscle wasting only
C. No fatty liver
D. Low insulin
33. Marasmus is characterized by:
A. Edema
B. Severe wasting
C. Fatty liver
D. Hypoalbuminemia only
34. During starvation, muscle releases:
A. Leucine and lysine
B. Alanine and glutamine
C. Phenylalanine and tyrosine
D. Methionine and cysteine
35. The major amino acid for ammonium trapping in the kidney is:
A. Alanine
B. Glycine
C. Glutamine
D. Serine
36. Essential amino acids are:
A. Alanine, glycine, serine
B. Leucine, valine, lysine
C. Tyrosine, cysteine
D. Proline, arginine in adults
37. Tyrosine is synthesized from:
A. Leucine
B. Glycine
C. Phenylalanine
D. Valine
38. Dopa decarboxylase requires:
A. PLP
B. THF
C. Biotin
D. FAD
39. Amino acids important for one-carbon metabolism include:
A. Glycine & serine
B. Leucine & tryptophan
C. Tyrosine & phenylalanine
D. Arginine & lysine
40. Homocysteine is formed from:
A. Serine
B. Methionine
C. Lysine
D. Glycine
41. SAM (S-adenosylmethionine) is:
A. Methyl donor
B. Biotin carrier
C. Antioxidant
D. Precursor of urea
42. Creatine is synthesized from:
A. Glycine + Arginine
B. Glycine + Lysine
C. Methionine + Tyrosine
D. Alanine + Glutamine
43. Major amino acid in collagen is:
A. Valine
B. Serine
C. Glycine
D. Glutamate
44. Hydroxylation of proline requires:
A. Vit B6
B. Vit C
C. Vit K
D. FAD
45. Nitric oxide is formed from:
A. Glycine
B. Arginine
C. Proline
D. Serine
46. GABA is synthesized from:
A. Tyrosine
B. Serine
C. Glutamate
D. Glycine
47. Which amino acid is purely ketogenic?
A. Isoleucine
B. Phenylalanine
C. Tyrosine
D. Leucine
48. Glucose-alanine cycle transfers:
A. CO₂
B. Ammonia to liver
C. Ketones
D. Fatty acids
49. Which amino acid forms histamine?
A. Histidine
B. Arginine
C. Tryptophan
D. Tyrosine
50. Rate of protein turnover is highest in:
A. Bone
B. Skin
C. Intestinal mucosa
D. Muscle
✅ ANSWER KEY
1-B
2-B
3-B
4-B
5-C
6-B
7-B
8-C
9-A
10-C
11-D
12-B
13-B
14-C
15-B
16-C
17-C
18-C
19-A
20-B
21-C
22-B
23-C
24-C
25-C
26-B
27-C
28-A
29-D
30-B
31-C
32-A
33-B
34-B
35-C
36-B
37-C
38-A
39-A
40-B
41-A
42-A
43-C
44-B
45-B
46-C
47-D
48-B
49-A
50-C