Introduction
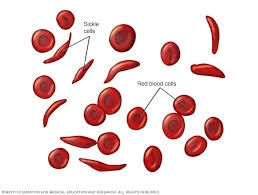
- Sickle cell anemia is a serious genetic disorder of the blood where the red blood cells (RBCs) become abnormally shaped.
- Instead of being round and flexible, like a donut, the cells take on a crescent or sickle shape.
- This change happens because of a problem with the hemoglobin inside the RBCs, specifically hemoglobin S (HbS).
- The sickle-shaped cells don’t move easily through blood vessels, which can block blood flow, leading to pain and damage in organs like the brain, lungs, and kidneys.
- This disorder is inherited, meaning you are born with it if both of your parents pass down the sickle cell gene.
- It’s most common in people of African, Mediterranean, Middle Eastern, and Indian descent.
- While sickle cell anemia can’t be cured by most treatments, people with the disease can manage their symptoms and live productive lives with proper care.
Etiology (Causes)
Sickle cell anemia is caused by a genetic mutation. A mutation in the gene responsible for making hemoglobin leads to the production of hemoglobin S (HbS) instead of normal hemoglobin A (HbA). This mutated hemoglobin doesn’t work properly, and when it loses oxygen, it sticks together and forms long, stiff rods inside the red blood cells. These rods cause the cells to take on a sickle shape, which makes them rigid and sticky.
-
Inheritance Pattern:
-
Sickle cell anemia is passed down in an autosomal recessive pattern.
-
This means that a person needs to inherit two copies of the sickle cell gene—one from each parent—to develop the disease.
-
If only one sickle cell gene is inherited, the person is a carrier, known as having sickle cell trait.
-
Carriers usually don’t show symptoms of the disease but can pass the gene to their children.
-
-
Co-inheritance of Other Genes:
-
Sometimes, people may have sickle cell disease alongside another genetic disorder, such as thalassemia, which can affect how severe their symptoms are.
-
The more genetic factors involved, the more complex the condition can become.
-
Epidemiology
Sickle cell anemia affects millions of people worldwide, with the highest numbers in regions with historical exposure to malaria. This is because having sickle cell trait can protect people from severe malaria, which is why the gene is common in malaria-prone regions.
-
Sub-Saharan Africa: The most common region for sickle cell disease. It is estimated that about 1 in 365 African American babies born in the U.S. have sickle cell anemia.
-
Middle East, Mediterranean, and India: These regions also have significant cases, but the frequency is not as high as in Africa. People of Mediterranean or Indian descent might also inherit sickle cell disease.
-
Worldwide: Approximately 300,000 children are born with sickle cell anemia each year. In the U.S., sickle cell anemia is most common in African Americans but is also found in other groups such as Hispanics, people from the Mediterranean, and Middle Eastern populations.
Pathophysiology
The pathophysiology of sickle cell anemia refers to the series of events that happen inside the body when someone has the disease. The major problem arises from how the red blood cells behave when hemoglobin S is present.
-
Sickle Cell Formation:
-
When the oxygen level in the blood drops, hemoglobin S molecules start sticking together.
-
This causes the red blood cells to change shape, becoming rigid and sickle-like instead of flexible, round, and smooth.
-
These sickle-shaped cells can’t pass easily through small blood vessels, which can block blood flow.
-
-
Hemolysis (Destruction of Red Blood Cells):
-
Normal red blood cells live for about 120 days.
-
But sickle cells are fragile and break down more quickly.
-
The premature destruction of sickle cells leads to anemia, where the body does not have enough healthy red blood cells to carry oxygen.
-
-
Vaso-occlusion (Blockage of Blood Flow):
-
The sickle cells can block blood vessels, leading to poor blood circulation.
-
This causes pain, known as a pain crisis, and can result in damage to organs and tissues due to the lack of oxygen.
-
When blood flow is blocked, it can cause swelling, infection, or even permanent damage to organs like the kidneys, heart, and brain.
-
-
Inflammation:
-
Sickle cells can cause inflammation in the blood vessels, which can lead to further blocking and tissue damage.
-
Over time, inflammation can make the blood vessels stiffer, making it harder for the body to repair itself.
-
-
Organ Damage:
-
Chronic pain, poor blood flow, and organ damage due to vaso-occlusion lead to long-term issues.
-
For example, the spleen, which helps filter the blood and fight infections, can become damaged over time (a condition known as autosplenectomy).
-
This makes people with sickle cell more vulnerable to infections.
-
Laboratory Investigations
Laboratory investigations are essential for diagnosing sickle cell anemia, monitoring its progression, and evaluating complications. These tests help healthcare providers understand the severity of the disease and guide treatment decisions. Below are the most commonly used tests for diagnosing and managing sickle cell anemia:
Complete Blood Count (CBC)
The Complete Blood Count (CBC) is one of the most basic and informative tests used to assess the overall health of a person and to help diagnose various blood disorders, including sickle cell anemia.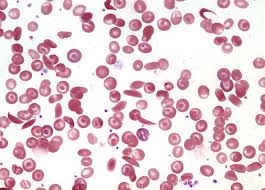
-
Red Blood Cell (RBC) Count: People with sickle cell anemia typically have a low RBC count due to the increased destruction of sickle-shaped red blood cells (hemolysis). This leads to anemia, where the blood lacks enough healthy RBCs to carry oxygen efficiently.
-
Reticulocyte Count: The reticulocyte count, which measures the number of immature red blood cells being produced by the bone marrow, is usually elevated in individuals with sickle cell anemia. This indicates that the body is attempting to compensate for the premature destruction of red blood cells by increasing production of new ones.
-
Hemoglobin Level: The hemoglobin concentration in the blood is typically low in sickle cell anemia, contributing to anemia.
-
White Blood Cell (WBC) Count: A mild increase in WBC count may be seen, particularly during pain crises or infections, as the body responds to stress and inflammation.
-
Platelet Count: Platelets may be elevated in sickle cell anemia, often due to the increased inflammation and tissue damage caused by sickled cells.
Peripheral Blood Smear
A peripheral blood smear is a test where a sample of blood is spread on a slide and examined under a microscope. This test is crucial for visually identifying abnormal red blood cells and other signs that help diagnose sickle cell anemia.
-
Sickle-Shaped Cells: The most distinctive feature seen on a blood smear in sickle cell anemia is the presence of sickle-shaped red blood cells. These cells are crescent-shaped rather than round and flexible, which makes them less able to flow through blood vessels efficiently.
-
Target Cells: These are RBCs that have a dark center surrounded by a lighter ring, often seen in sickle cell anemia. Target cells are typically a result of abnormal cell membrane structure and are common in hemoglobinopathies.
-
Howell-Jolly Bodies: These are remnants of the cell nucleus, seen in patients with a dysfunctional or absent spleen (as seen in sickle cell disease). The spleen is often damaged over time due to repeated blockages of blood vessels.
-
Nucleated Red Blood Cells: Occasionally, nucleated RBCs may be seen in sickle cell anemia. This usually indicates a more severe form of anemia and is a sign that the bone marrow is rapidly producing new RBCs to compensate for the loss of sickled cells.
Hemoglobin Electrophoresis
Hemoglobin electrophoresis is the gold standard test for diagnosing sickle cell anemia and is used to identify the different types of hemoglobin present in a person’s blood. It works by separating different types of hemoglobin based on their movement in an electric field.
-
Hemoglobin S (HbS): In sickle cell anemia, hemoglobin S is the dominant hemoglobin present. This test will show a significant amount of HbS and very little, or no, hemoglobin A (HbA).
-
Hemoglobin F (HbF): In some individuals, particularly those who have received treatment with hydroxyurea, an increased amount of fetal hemoglobin (HbF) may be present. HbF is protective against sickling, and its higher levels can reduce the frequency of painful crises.
-
Hemoglobin A: In individuals with sickle cell anemia, there should be little to no hemoglobin A detected because their red blood cells are mostly composed of hemoglobin S. However, a person with sickle cell trait (carrier) will have a mixture of HbS and HbA, with HbA being dominant.
-
Hemoglobin C or Other Abnormal Hemoglobins: This test also helps identify other hemoglobinopathies that may be coexisting with sickle cell disease, such as hemoglobin C disease or hemoglobin SC disease, which can influence the disease’s severity.
Sickle Cell Solubility Test
The sickle cell solubility test is a screening test that detects the presence of hemoglobin S (HbS) in the blood. It is less specific than hemoglobin electrophoresis and is often used as an initial test in areas where sickle cell anemia is common.
-
The test involves adding a reagent to a blood sample that causes the hemoglobin S molecules to precipitate (form solid clumps) when oxygen levels drop. If sickle cell hemoglobin is present, the solution will become turbid (cloudy), indicating a positive result.
-
This test is quick and simple but cannot differentiate between sickle cell anemia (two copies of the sickle cell gene) and sickle cell trait (one copy of the sickle cell gene). Therefore, it is typically followed up by a more detailed test, such as hemoglobin electrophoresis.
Reticulocyte Count
The reticulocyte count measures the number of young, immature red blood cells being released into the bloodstream from the bone marrow. In sickle cell anemia, the reticulocyte count is usually elevated as the bone marrow works overtime to replace the red blood cells destroyed by the disease.
-
Increased Reticulocytes: A high reticulocyte count suggests that the body is compensating for the loss of red blood cells by increasing production. This is often observed in the setting of an acute hemolytic crisis when sickle cells are destroyed rapidly.
-
Chronic Anemia: Despite an increased reticulocyte count, sickle cell anemia is a chronic form of anemia, and the total red blood cell count remains low due to ongoing destruction of sickle cells.
Lactate Dehydrogenase (LDH) and Bilirubin Levels
Lactate dehydrogenase (LDH) and bilirubin levels are used to assess the extent of RBC destruction (hemolysis) in sickle cell anemia.
-
LDH: LDH is an enzyme found in red blood cells. When RBCs break down prematurely, LDH is released into the bloodstream. Elevated LDH levels indicate increased hemolysis and tissue damage. A significant increase in LDH can also suggest organ damage, such as in the liver or kidneys.
-
Bilirubin: Bilirubin is produced when hemoglobin is broken down. Increased hemolysis results in high bilirubin levels, particularly unconjugated bilirubin, which can lead to jaundice (yellowing of the skin and eyes). In sickle cell anemia, chronic hemolysis often leads to persistent mild jaundice.
Doppler Ultrasound and MRI
Doppler ultrasound and MRI are imaging techniques that help monitor complications of sickle cell anemia, especially those involving the brain and other organs.
-
Doppler Ultrasound of the Cerebral Arteries: In children with sickle cell anemia, a Doppler ultrasound is used to assess blood flow in the brain’s major arteries. This is crucial for detecting early signs of stroke or cerebrovascular disease. Regular screening with Doppler ultrasound can help identify patients at high risk of stroke, allowing for early intervention.
-
MRI of the Brain: MRI is particularly useful for detecting silent strokes (strokes that do not show obvious symptoms) and other brain abnormalities caused by sickle cell disease. MRI can also help monitor the effects of vaso-occlusive crises on the brain and other organs.
-
MRI of Other Organs: MRI can also be used to assess the health of other organs, such as the liver, spleen, and kidneys, to detect signs of organ damage from repeated vaso-occlusion or hemolysis.
Chest X-Ray
A chest X-ray is used to identify respiratory complications that are common in sickle cell anemia, such as acute chest syndrome.
-
Acute Chest Syndrome: This is a life-threatening complication of sickle cell anemia that results from lung tissue damage caused by blocked blood vessels. Chest X-rays may show areas of lung consolidation, suggesting pneumonia or infarction (tissue death due to lack of blood flow).
-
Other Lung Abnormalities: Chronic lung damage in sickle cell anemia may also lead to changes in the lungs visible on chest X-rays.
Treatment and Management
There are several ways to treat and manage sickle cell anemia. While there is no universal cure, treatments aim to reduce symptoms and prevent complications.
-
Pain Management: Pain from sickle cell crises can be severe. Treatments include:
-
Pain relief medications such as opioids or NSAIDs (non-steroidal anti-inflammatory drugs).
-
Hydration: Drinking enough fluids helps keep the blood less sticky.
-
Oxygen therapy: Used during crises to increase the oxygen in the blood, reducing the chance of cells sickling.
-
-
Blood Transfusions: In severe cases, regular blood transfusions may be used to replace sickled cells with normal red blood cells. This reduces the risk of strokes and other complications.
-
Hydroxyurea: This drug can increase the production of fetal hemoglobin (HbF), which prevents red blood cells from sickling. It is commonly used to reduce pain crises and the need for blood transfusions.
-
Bone Marrow or Stem Cell Transplantation: The only potential cure for sickle cell anemia is a bone marrow transplant from a compatible donor. However, this is a complex procedure with significant risks.
-
Antibiotics and Vaccinations: People with sickle cell anemia are at a higher risk of infection, especially from bacteria like Streptococcus pneumoniae. Routine vaccinations and sometimes antibiotics, particularly penicillin, are given from a young age to prevent infections.
-
Gene Therapy: New treatments are being developed that aim to change the faulty gene that causes sickle cell anemia. This is a rapidly advancing area of research.
Management
Managing sickle cell anemia involves keeping an eye on potential complications and addressing them early:
-
Prevention of Complications: Regular doctor visits and staying up-to-date on treatments like blood transfusions, medications, and vaccines are key to preventing complications such as stroke, kidney failure, or organ damage.
-
Psychosocial Support: Dealing with chronic pain and the uncertainty of disease progression can be emotionally difficult. Counseling and mental health support are important for patients and their families.
-
Education: Patients and caregivers need education about the disease, recognizing signs of pain crises, staying hydrated, and knowing when to seek medical care. Awareness of genetic risks can help with family planning.
-
Prenatal Screening: For couples with a family history of sickle cell anemia, genetic counseling and prenatal testing can help identify the risk of having a child with the disease.