Introduction
- Thalassemia is a group of inherited blood disorders characterised by reduced or absent production of one of the globin chains of haemoglobin, resulting in anaemia.
- Haemoglobin is a protein found in red blood cells that is responsible for transporting oxygen throughout the body.
- When thalassemia occurs, there is either insufficient production of alpha-globin or beta-globin chains, depending on the type of thalassemia.
-
Alpha-thalassemia occurs when there is a defect in the genes that produce the alpha-globin chains.
-
Beta-thalassemia occurs when there is a defect in the genes that produce the beta-globin chains.
- The condition results in two major types: thalassemia minor (carrier status with mild symptoms) and thalassemia major (severe disease requiring intensive medical management).
Aetiology
The root cause of thalassemia lies in genetic mutations in the alpha-globin and beta-globin genes.
-
Alpha-Thalassemia:
-
Caused by deletions or mutations in the HBA1 and HBA2 genes located on chromosome 16. These genes are responsible for producing the alpha-globin chains of hemoglobin.
-
Alpha-thalassemia trait (carriers) occurs when one of the alpha-globin genes is deleted or mutated, resulting in mild anemia or no symptoms at all.
-
Alpha-thalassemia major (hydrops fetalis) is seen when all four alpha-globin genes are defective. This condition results in a fatal form of anemia in utero, and is typically diagnosed by prenatal genetic testing.
-
-
Beta-Thalassemia:
-
Caused by mutations in the HBB gene located on chromosome 11, which codes for the beta-globin chains of hemoglobin.
-
Beta-thalassemia minor (also known as thalassemia trait) occurs when a person inherits one defective beta-globin gene, typically resulting in mild anemia with few or no symptoms.
-
Beta-thalassemia major (Cooley’s anemia) occurs when a person inherits two defective beta-globin genes. This leads to severe anemia, requiring regular blood transfusions.
-
Inheritance is autosomal recessive, meaning both parents must carry a defective gene for their child to have the disease. If one parent is a carrier, the child may inherit the trait but usually not the disease.
Epidemiology
Thalassemia is a global health concern, with certain regions of the world having higher prevalence rates due to genetic factors. Its epidemiology includes:
-
Geographic Distribution:
-
Beta-thalassemia is most common in populations of Mediterranean descent, such as Greeks, Italians, and Arabs. It is also prevalent in South Asia, Southeast Asia, and parts of North Africa.
-
Alpha-thalassemia is most commonly found in populations from Southeast Asia, China, and Africa.
-
Countries like India, Pakistan, Bangladesh, Thailand, and China have high rates of thalassemia, with some estimates suggesting that 5-7% of people in these regions may carry a thalassemia gene.
-
-
Global Prevalence:
-
Thalassemia is one of the most common genetic disorders worldwide, with millions of carriers and hundreds of thousands of affected individuals.
-
Thalassemia major (severe disease) often manifests early in life, making it a major cause of morbidity and mortality in affected regions.
-
-
Carrier Status:
-
Around 5% of the global population are carriers of the thalassemia gene, which may go unnoticed because carriers typically exhibit only mild or no symptoms.
-
-
Impact on Health Systems:
-
In regions with a high prevalence, thalassemia imposes a significant burden on healthcare systems, especially in terms of blood transfusion programs and iron chelation therapy.
-
Pathophysiology
The pathophysiology of thalassemia is complex, and it involves two key processes: ineffective erythropoiesis and hemolysis. These result in anemia, which is the hallmark feature of the disease.
-
Imbalanced Hemoglobin Production:
-
Normal hemoglobin (Hb) is made of two alpha-globin and two beta-globin chains. In thalassemia, the production of one type of globin chain (either alpha or beta) is reduced or absent.
-
In beta-thalassemia, the beta-globin chains are underproduced or absent, while the production of alpha-globin chains continues at a normal or higher level.
-
In alpha-thalassemia, the alpha-globin chains are underproduced or absent, leading to an excess of beta-globin chains.
-
-
Excess of Unpaired Chains:
-
The unpaired globin chains (either excess alpha or beta chains) are unstable and precipitate inside the red blood cells. This results in the destruction of these cells (hemolysis) both in the bone marrow (ineffective erythropoiesis) and in the peripheral circulation.
-
-
Bone Marrow Expansion:
-
The body attempts to compensate for the anemia by increasing the production of red blood cells in the bone marrow, leading to an expansion of the marrow and skeletal deformities, particularly in the face and skull (known as “chipmunk facies”).
-
-
Iron Overload:
-
Frequent blood transfusions to treat thalassemia can lead to excess iron in the body, which is deposited in organs such as the liver, heart, and endocrine glands, leading to iron overload. This can cause serious complications like liver failure, heart failure, and diabetes.
-
Laboratory Investigations
Diagnosis and monitoring of thalassemia require a series of laboratory tests:
-
Complete Blood Count (CBC):
-
Microcytic hypochromic anemia: Characterized by smaller than normal red blood cells (low mean corpuscular volume, MCV) and pale cells (low mean corpuscular hemoglobin, MCH).
-
Low hemoglobin (Hb) levels.
-
Increased reticulocyte count indicating bone marrow response.
-
-
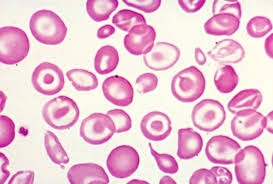
Peripheral Blood Smear:
-
Target cells (red blood cells with a dark center and a ring around it).
-
Nucleated red blood cells: Immature red blood cells found in the bloodstream.
-
Anisopoikilocytosis: Variation in red blood cell size and shape.
-
-
Hemoglobin Electrophoresis:
-
This test is crucial for diagnosing thalassemia and distinguishing between its subtypes.
-
In beta-thalassemia, there will be elevated HbA2 and HbF (fetal hemoglobin) levels.
-
In alpha-thalassemia, the hemoglobin electrophoresis is often normal, as the defect is primarily in alpha-globin chain production.
-
-
Genetic Testing:
-
DNA analysis can identify specific mutations in the HBA1, HBA2, and HBB genes. This is particularly important for prenatal diagnosis and carrier screening.
-
-
Iron Studies (Serum Ferritin, Serum Iron, Transferrin Saturation):
-
These tests monitor iron overload, a common complication in thalassemia patients receiving blood transfusions.
-
Elevated serum ferritin levels indicate iron overload.
-
-
Liver Function Tests:
-
Abnormal results may indicate iron buildup in the liver, causing liver damage.
-
-
*MRI (T2 for Iron Overload)**:
-
Cardiac and liver MRI can be used to quantify iron overload, helping to guide treatment with iron chelation.
-
Management
The management of thalassemia is aimed at controlling symptoms, preventing complications, and improving quality of life. Treatment approaches depend on the severity of the disease.
-
General Measures:
-
Folic acid supplementation is essential to support red blood cell production.
-
Regular monitoring of hemoglobin levels, iron status, and organ function is required.
-
-
Blood Transfusions:
-
The cornerstone of treatment for thalassemia major. Regular blood transfusions help maintain normal hemoglobin levels (usually >9-10 g/dL) and prevent severe anemia.
-
Transfusion therapy is typically required every 2-4 weeks.
-
-
Iron Chelation Therapy:
-
Frequent transfusions lead to excess iron in the body, which is toxic to organs. Iron chelation therapy is used to remove excess iron.
-
Common chelators include:
-
Deferoxamine: Administered via subcutaneous or intravenous infusion.
-
Deferasirox and Deferiprone: Oral chelators that are more convenient and effective.
-
-
Regular monitoring of iron levels is necessary to avoid complications from iron overload.
-
-
Bone Marrow/Stem Cell Transplantation:
-
The only curative treatment for thalassemia is hematopoietic stem cell transplantation (HSCT), or bone marrow transplant. The success rate is higher when performed in children before significant organ damage occurs, and when a matched sibling donor is available.
-
-
Gene Therapy:
-
Advances in gene therapy offer hope for curative treatments. Gene therapy aims to correct the genetic mutations responsible for thalassemia. Clinical trials are ongoing, and there have been some promising results with autologous stem cells modified to express functional beta-globin.
-
-
Splenectomy:
-
The spleen often enlarges due to increased red blood cell destruction. In cases where splenomegaly leads to complications like hypersplenism (increased destruction of blood cells), a splenectomy may be considered.
-
-
Prenatal Diagnosis and Genetic Counselling:
-
Prenatal genetic testing (amniocentesis or chorionic villus sampling) can diagnose thalassemia in the fetus, allowing parents to make informed decisions. Genetic counselling is crucial for couples at risk of having children with thalassemia.
-