Introduction
- Alkaptonuria is a rare inherited metabolic disorder characterized by the deficiency of the enzyme homogentisate 1,2-dioxygenase (HGD), leading to the accumulation of homogentisic acid (HGA) in the body.
- This accumulated acid is excreted in the urine and deposited in connective tissues, producing darkened urine and bluish-black pigmentation of cartilage and other tissues — a condition termed ochronosis.
- It is one of the earliest known inborn errors of metabolism, first described by Sir Archibald Garrod in 1902, who introduced the concept of “one gene–one enzyme” relationship using this disease as an example.
Biochemical Basis and Pathophysiology
Normal Pathway of Tyrosine Catabolism
Tyrosine, derived from phenylalanine, is degraded through a multistep pathway:
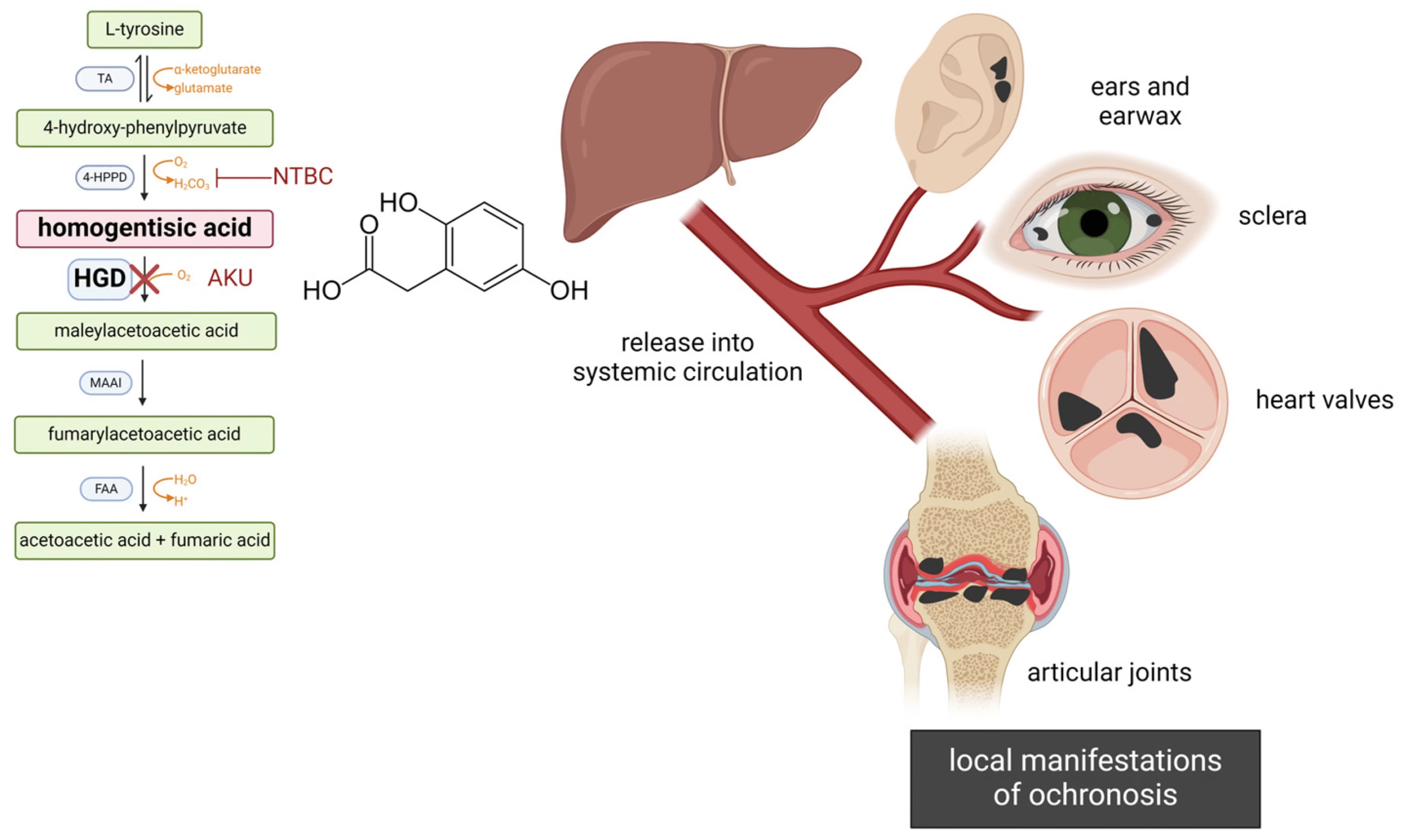
Phenylalanine → Tyrosine → p-Hydroxyphenylpyruvate → Homogentisic acid → Maleylacetoacetic acid → Fumarate + Acetoacetate
-
The conversion of homogentisic acid to maleylacetoacetic acid is catalyzed by homogentisate 1,2-dioxygenase (HGD), a hepatic cytosolic enzyme.
Defect in Alkaptonuria
-
In alkaptonuria, HGD enzyme deficiency leads to accumulation of homogentisic acid in body fluids and tissues.
-
HGA is oxidized spontaneously to benzoquinone acetic acid, which polymerizes to form melanin-like pigments that deposit in connective tissues — causing ochronosis.
Genetics
| Feature | Details |
|---|---|
| Gene involved | HGD gene (chromosome 3q13.33) |
| Inheritance | Autosomal recessive |
| Carrier frequency | 1 in 250–500 |
| Disease incidence | ~1 in 2,50,000–10,00,000 live births (higher in Slovakia and the Dominican Republic) |
| Mutation effect | Missense, nonsense, or frameshift mutations → defective or absent HGD enzyme |
A child develops the disease only if both parents are carriers of the mutant allele.
Pathophysiology of Pigment Deposition
-
Homogentisic acid accumulation in blood and tissues.
-
Oxidation of HGA → benzoquinone acetic acid → polymerization into ochronotic pigment.
-
Pigments deposit in cartilage, tendons, sclera, skin, and heart valves, causing:
-
Stiffness and brittleness of cartilage.
-
Dark bluish-black discoloration (especially in ear cartilage and sclera).
-
Degenerative arthritis and cardiac valvular calcification.
-
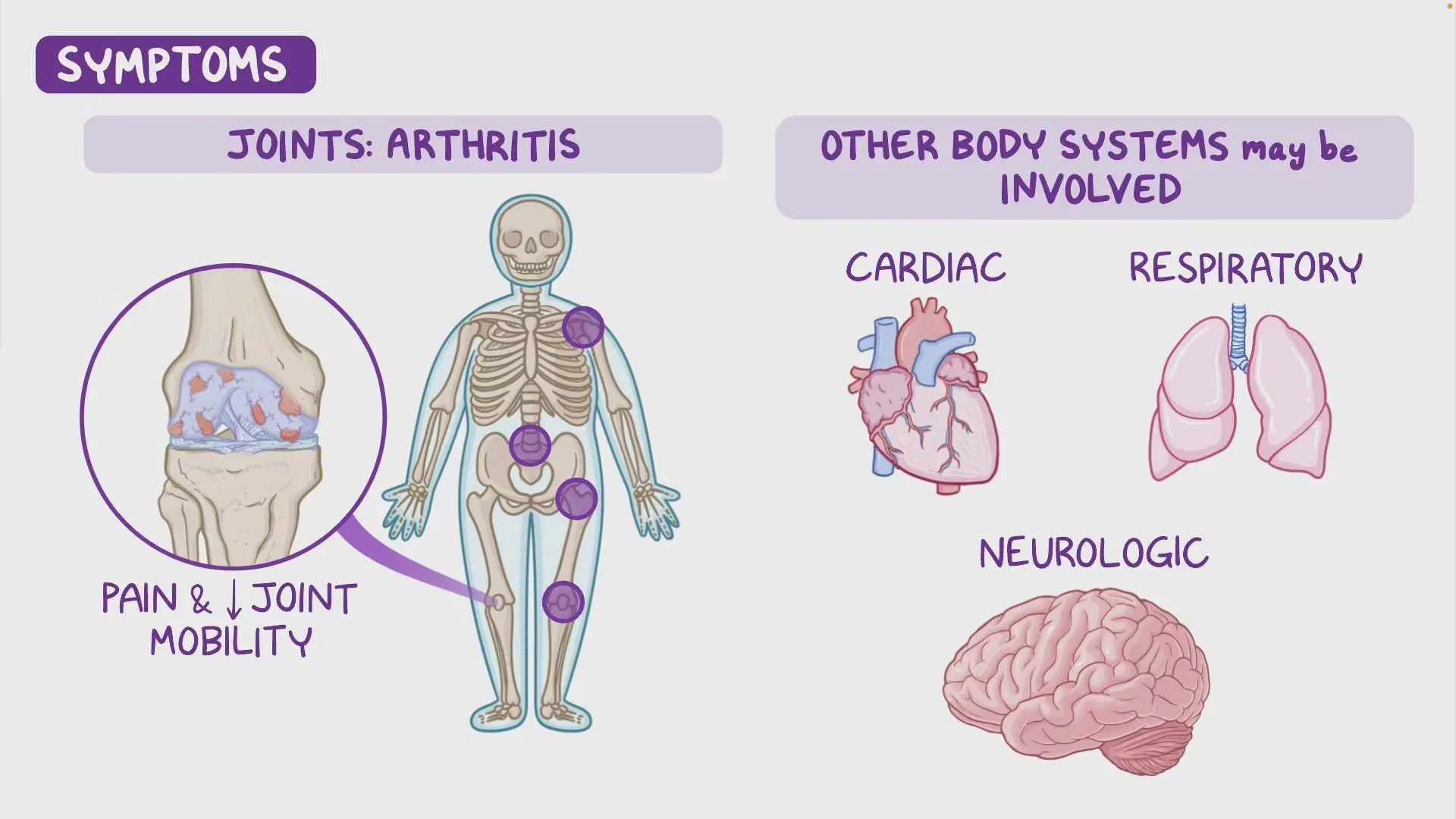
Clinical Features
| Age of Onset | Clinical Manifestations |
|---|---|
| Infancy | Urine darkens on standing (due to oxidation of HGA to black pigment) |
| Adulthood (30–40 yrs) | Arthropathy of large joints and spine; chronic low back pain |
| Later life | Ochronotic pigmentation of ears, sclera, and skin; cardiac and renal complications |
Major Features
-
Urine darkens to brown/black when exposed to air or alkali.
-
Ochronosis: Bluish-black pigmentation of connective tissues (ear cartilage, sclera, nails).
-
Arthritis: Degeneration of intervertebral discs and large joints (especially spine, knees, hips).
-
Cardiac involvement: Calcification and stenosis of aortic and mitral valves.
-
Renal/Prostatic stones: Due to HGA deposition and crystallization.
-
No mental retardation or growth retardation (unlike phenylketonuria).
Laboratory Diagnosis
1. Urine Examination
-
Darkening of urine on standing — diagnostic hallmark.
-
Black precipitate with NaOH or NH₄OH due to oxidation of HGA.
-
Benedict’s test: Positive (reducing property of HGA).
-
Ferric chloride test: Transient blue-green color.
-
Paper chromatography / TLC / HPLC: Detects elevated HGA levels.
2. Quantitative Estimation
-
Spectrophotometric or HPLC quantification of HGA in urine.
-
24-hour urinary excretion of HGA markedly increased (up to 1–8 g/day).
3. Molecular Diagnosis
-
HGD gene sequencing for mutation identification.
-
Carrier detection and prenatal diagnosis possible through genetic testing.
Differential Diagnosis
| Condition | Distinguishing Feature |
|---|---|
| Phenylketonuria | Elevated phenylalanine; mental retardation; fair complexion |
| Tyrosinemia type I | Liver and renal failure; succinylacetone in urine |
| Tyrosinemia type II | Corneal ulcers, keratitis |
| Melaninuria | Occurs in melanoma; no joint or cartilage involvement |
Complications
-
Progressive degenerative arthritis (particularly of spine and large joints).
-
Cardiac valvular disease (calcified aortic and mitral valves).
-
Nephrolithiasis / Prostatolithiasis (stones due to HGA precipitation).
-
Hearing loss due to pigment deposition in ear cartilage.
-
Restrictive chest movement due to costovertebral fusion in severe cases.
Treatment
A. Dietary Management
-
Low-protein diet restricting phenylalanine and tyrosine intake to reduce HGA formation.
-
Avoid meat, fish, cheese, and legumes (high in aromatic amino acids).
-
-
Ascorbic acid (Vitamin C) (500 mg to 1 g/day):
-
Acts as antioxidant; may slow ochronosis and pigment polymerization.
-
B. Pharmacological Therapy
-
Nitisinone (NTBC, 2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione):
-
Inhibits p-hydroxyphenylpyruvate dioxygenase (upstream enzyme).
-
Reduces HGA production by >90%.
-
Long-term therapy improves clinical outcomes; FDA-approved for hereditary tyrosinemia type I and under evaluation for alkaptonuria.
-
C. Supportive Treatment
-
Analgesics and physiotherapy for arthropathy.
-
Joint replacement surgery in advanced arthritis.
-
Cardiac surgery for severe valvular calcification.
-
Adequate hydration to reduce renal stone risk.
D. Gene Therapy (Future Prospect)
-
Experimental studies explore HGD gene replacement using recombinant viral vectors.
Epidemiology
-
Global incidence: ~1 in 2.5–10 lakh births.
-
High prevalence in Slovakia, Dominican Republic, and some regions of India (due to founder mutations and consanguinity).
Historical Perspective
-
Sir Archibald Garrod (1902) identified alkaptonuria as the first inborn error of metabolism.
-
Demonstrated that metabolic disorders could result from enzyme defects caused by gene mutations, pioneering biochemical genetics.