Determination of abnormal hemoglobin by various methods
- Electrophoresis is based on the principle of charge-to-mass ratio of molecules.
- Hemoglobin molecules are proteins that carry a charge, and when an electric field is applied to a sample, the hemoglobin molecules migrate towards the electrode opposite to their charge.
- Different types of hemoglobin (such as HbA, HbS, HbC) have different electrical charges and thus will migrate at different rates on an agarose gel.
- The mobility depends on the net charge of the hemoglobin molecule and its size.
- Abnormal hemoglobins, such as HbS (sickle cell hemoglobin) or HbC, differ in their charge and migration pattern compared to normal HbA, allowing for their identification and separation.
Hemoglobin Solubility Test (for HbS Detection)
Principle:
- The solubility test is based on the fact that deoxygenated hemoglobin S (HbS) forms insoluble polymers when exposed to low oxygen conditions.
- These polymers precipitate out of solution, causing the solution to become turbid.
- In contrast, normal hemoglobin (HbA) remains soluble even under deoxygenated conditions.
- The presence of turbidity (cloudiness) in the solution indicates the presence of HbS (sickle hemoglobin).
- This test is commonly used as a quick screening method for sickle cell disease (SCD).
Chromatography (HPLC)
Principle:
- High-Performance Liquid Chromatography (HPLC) is based on the principle of differential partitioning of molecules between a stationary phase (column) and a mobile phase (solvent).
- Hemoglobin molecules, due to their differing chemical compositions (amino acid sequences, charge, and structure), interact differently with the stationary phase, causing them to be separated as they pass through the column.
- Each type of hemoglobin (such as HbA, HbS, HbF, HbC) has a unique retention time because of its distinct chemical properties.
- As the hemoglobin molecules pass through the column, they are detected and identified by their characteristic retention time and peak formation in the chromatogram.
- This method allows for the precise quantification and identification of various hemoglobin types in a blood sample.
Materials and Equipment
-
Blood sample (whole blood or blood smear)
Used as the source to extract hemoglobin. The sample can be collected from a patient or donor. -
Hemoglobin electrophoresis kit
This kit typically includes an agarose gel, buffer solution, and electrodes for performing the electrophoresis test. -
Hemoglobin solubility test kit (e.g., sodium dithionite test)
This test is designed to detect sickle cell disease by checking the solubility of deoxygenated hemoglobin S (HbS). -
Chromatography equipment
You can use either Thin-Layer Chromatography (TLC) or High-Performance Liquid Chromatography (HPLC). The chromatography system includes a column, mobile phase, and detector (UV or fluorescence). -
Microhematocrit tubes for centrifuge
Used to separate blood components in some preparatory procedures, if needed. -
Centrifuge
A machine used to spin the blood sample to separate components, often used in preparatory steps. -
Pipettes and test tubes
For transferring liquids and handling samples. -
Distilled water
Used for diluting or preparing solutions for various tests. -
Sodium citrate solution
Used for anticoagulating blood samples to prevent clotting, ensuring the hemoglobin remains in its liquid form for testing. -
pH buffer solutions
These are necessary to maintain the required pH levels during electrophoresis and chromatography to ensure proper separation of hemoglobin molecules. -
Heat block or water bath (for solubility test)
Used to control temperature during the solubility test to maintain optimal conditions for the reaction. -
Reagents:
-
Sodium dithionite solution for solubility testing, which reduces hemoglobin, promoting the precipitation of HbS.
-
Hemoglobin standards like HbA, HbS, and HbC for comparison in electrophoresis or chromatography.
-
Agarose gel for electrophoresis to separate the hemoglobin variants.
-
Electrophoresis buffer (usually an alkaline buffer with a pH of 8.6) for proper mobility of hemoglobins during electrophoresis.
-
Chromatography solvent for HPLC, including water, acetonitrile, and a pH-adjusted buffer.
-
Procedure
Hemoglobin Electrophoresis:
This is the most widely used method for separating different hemoglobin types.
-
Sample Preparation:
-
Collect a fresh blood sample (usually 2-5 mL of venous blood).
-
Dilute it in sodium citrate solution (to prevent clotting) and centrifuge if needed to obtain plasma or red cell suspension.
-
-
Gel Setup:
-
Prepare an agarose gel in a specialized electrophoresis tray. The gel has tiny pores that help separate different hemoglobin variants by their size and charge.
-
Prepare an electrophoresis buffer solution with a pH of 8.6 (typically alkaline) to ensure proper migration of the hemoglobin molecules.
-
Load the diluted blood sample into small wells on the gel, along with known hemoglobin standards (such as HbA, HbS, and HbC).
-
-
Electrophoresis:
-
Apply a constant electric current (typically 80-150 volts) for about 30-60 minutes.
-
The hemoglobin samples will migrate towards the anode (positive pole) of the electrophoresis chamber. The speed of migration depends on the net charge of the hemoglobin molecules. HbA, being negatively charged, migrates towards the positive electrode.
-
-
Staining and Visualization:
-
After electrophoresis, stain the gel with a hemoglobin-specific stain (e.g., Ponceau S, Amido Black).
-
Allow the gel to dry and then examine it under appropriate light.
-
Analyze the gel. Normal hemoglobin (HbA) will appear as a single band at a specific position. Abnormal hemoglobins (e.g., HbS, HbC) will form additional or shifted bands due to their altered charge.
-
-
Results:
-
Compare the position of the patient’s bands with known standards.
-
Normal: Only one band (HbA).
-
Abnormal: Additional bands (HbS, HbC, or a combination), indicating a hemoglobinopathy.
-
Hemoglobin Solubility Test (for HbS detection):
This simple, rapid test screens for sickle cell anemia.
-
Sample Preparation:
-
Collect the blood sample and transfer it into a clean test tube.
-
-
Test Solution:
-
Add a few drops of sodium dithionite solution. This chemical agent deoxygenates the hemoglobin, causing sickle hemoglobin (HbS) to polymerize and form insoluble aggregates.
-
-
Incubation:
-
After mixing, allow the sample to sit at room temperature for 10 minutes. This gives the hemoglobin enough time to deoxygenate and precipitate if HbS is present.
-
-
Observation:
-
Examine the test tube. If the solution turns cloudy or turbid, it indicates the presence of sickle hemoglobin, which precipitates when deoxygenated.
-
-
Interpretation:
-
Positive result: Cloudy solution, indicating the presence of HbS.
-
Negative result: Clear solution, indicating normal hemoglobin (HbA).
-
Chromatography (HPLC):
HPLC offers a highly sensitive and quantitative method for hemoglobin analysis.
-
Sample Preparation:
-
Dilute the blood sample into the sample preparation solution suitable for HPLC analysis.
-
-
Column Setup:
-
Install a chromatographic column (typically C18 or similar) into the HPLC machine. This column separates the components based on their interaction with the stationary phase.
-
Prepare the mobile phase, which often contains a mixture of acetonitrile, water, and a buffer, adjusted to a pH that promotes the separation of hemoglobin.
-
-
Separation Process:
-
Inject the prepared blood sample into the HPLC machine. Hemoglobin molecules will interact differently with the stationary phase and will elute from the column at different times.
-
Each hemoglobin type (e.g., HbA, HbS, HbF) has a characteristic retention time.
-
-
Detection:
-
The sample is passed through a UV or fluorescence detector that identifies the different hemoglobin variants based on their absorbance or fluorescence.
-
-
Results:
-
The chromatogram generated will show peaks at different retention times corresponding to each hemoglobin type.
-
Normal hemoglobin (HbA): A single peak.
-
Abnormal hemoglobins (e.g., HbS): A different retention time, resulting in a separate peak.
-
Clinical Significance
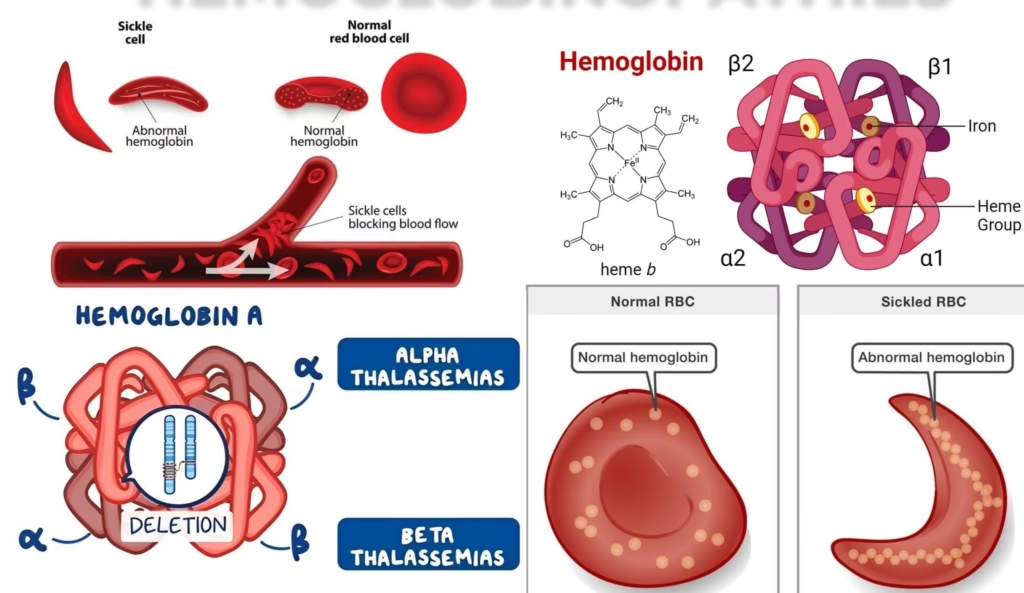
Sickle Cell Disease (HbS)
-
Clinical Significance:
-
Sickle cell disease (SCD) is one of the most common genetic disorders worldwide, particularly prevalent in regions such as sub-Saharan Africa, the Mediterranean, the Middle East, and India.
-
It is caused by a mutation in the beta-globin gene, leading to the production of hemoglobin S (HbS).
-
This hemoglobin variant forms sickle-shaped red blood cells under low oxygen conditions.
-
The sickled cells cause blockages in small blood vessels, leading to pain episodes, organ damage, and increased risk of infections.
-
-
Clinical Impact:
-
Pain crises: Episodes of severe pain due to vaso-occlusion in blood vessels.
-
Increased risk of stroke: Especially in children.
-
Organ damage: Over time, sickle cells can damage the spleen, liver, kidneys, and other organs.
-
Infections: Patients are more prone to bacterial infections, especially from encapsulated organisms like Streptococcus pneumoniae.
-
-
Management:
Early detection allows for better management of the disease, including pain control, preventive antibiotics, blood transfusions, and bone marrow transplants for certain patients.
Hemoglobin C Disease
-
Clinical Significance:
-
Hemoglobin C disease is caused by a mutation in the beta-globin gene, resulting in hemoglobin C (HbC).
-
The mutation causes the hemoglobin molecules to crystallize in red blood cells, leading to hemolysis (destruction of red blood cells) and mild anemia.
-
HbC disease is generally less severe than sickle cell disease, but it can still cause splenomegaly (enlarged spleen), mild jaundice, and fatigue.
-
-
Clinical Impact:
-
Mild hemolytic anemia: Chronic anemia can cause fatigue and pallor.
-
Splenomegaly: The spleen becomes enlarged as it works harder to remove damaged red blood cells.
-
Jaundice: Yellowing of the skin or eyes due to increased breakdown of red blood cells.
-
-
Management:
HbC disease usually requires supportive care. Management is generally focused on treating anemia and monitoring for any complications like gallstones, which can occur due to excessive breakdown of red blood cells.
Hemoglobin E Disease
-
Clinical Significance:
Hemoglobin E is another beta-globin mutation found predominantly in Southeast Asia. It is one of the most common hemoglobinopathies in this region. In HbE disease, hemoglobin E (HbE) causes mild anemia, but usually no severe clinical symptoms, although it can cause issues when combined with other hemoglobinopathies like thalassemia. -
Clinical Impact:
-
Mild anemia: The individual may have fatigue and paleness.
-
Complications when combined with thalassemia: In cases where an individual inherits HbE from one parent and thalassemia from the other, there can be severe thalassemia syndromes.
-
-
Management:
In the majority of cases, HbE disease does not require treatment. However, for individuals with co-inherited thalassemia, blood transfusions and possibly iron chelation therapy may be necessary.
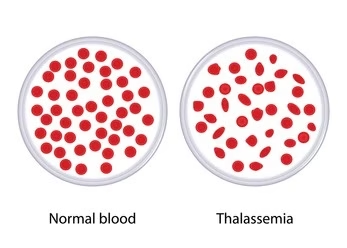
Thalassemia (Alpha and Beta Thalassemia)
-
Clinical Significance:
Thalassemia is a group of inherited blood disorders characterized by insufficient hemoglobin production. There are two main types:-
Alpha thalassemia: Caused by a deficiency in alpha-globin chains.
-
Beta thalassemia: Caused by a deficiency in beta-globin chains.
Clinical Impact:
-
Severe anemia: Patients often require regular blood transfusions.
-
Iron overload: Due to frequent transfusions, iron chelation therapy is necessary to prevent organ damage from excess iron.
-
Splenomegaly: The spleen may become enlarged as it works to remove abnormal red blood cells.
-
Growth retardation: Thalassemia can affect growth and development in children.
-
Bone deformities: Severe anemia can lead to bone marrow expansion, causing deformities.
-
-
Management:
Management includes blood transfusions, iron chelation, and sometimes bone marrow transplant. Gene therapy is being explored as a potential treatment in the future.
Fetal Hemoglobin (HbF) Persistence
-
Clinical Significance:
In some individuals, fetal hemoglobin (HbF) persists into adulthood. This may occur naturally in thalassemia or sickle cell disease, as HbF can reduce the severity of these conditions by inhibiting sickling or promoting better oxygen delivery. -
Clinical Impact:
-
Reduced symptoms: In individuals with sickle cell disease or thalassemia, elevated HbF levels are associated with a milder clinical course.
-
-
Management:
Research into inducing HbF production through medications like hydroxyurea is ongoing to treat sickle cell disease.