Introduction
- Plasma proteins are a heterogeneous group found in blood plasma, constituting a significant blood component.
- They originate primarily from the liver, with a few exceptions produced by immune cells and other tissues.
- Plasma proteins are crucial for various physiological processes, including maintaining oncotic pressure, facilitating immune responses, and transporting hormones and nutrients.
- The concentration of plasma proteins in normal human plasma is approximately 6-8 g/dL.
Classification
Plasma proteins can be classified based on various criteria, such as function, solubility, and electrophoretic mobility.
- By Function:
- Transport Proteins: Carry molecules like lipids, hormones, and metal ions.
- Examples:- Albumin (fatty acids, hormones), Transferrin (iron transport)
- Immunoglobulins: Play roles in the immune response.
- Examples:- IgG, IgA, IgM, IgE, IgD (different classes of antibodies)
- Clotting Factors: Involved in blood coagulation.
- Example:- Fibrinogen (converts to fibrin)
- Enzymatic Proteins: Facilitate biochemical reactions.
- Example:- Certain enzymes like lipoprotein lipase.
- Transport Proteins: Carry molecules like lipids, hormones, and metal ions.
- By Solubility:
- Soluble Proteins: Most plasma proteins, including albumin and globulins.
- Insulin and membrane-bound proteins: Less relevant in plasma context.
- By Electrophoretic Mobility:
- Albumin: Travels the farthest due to its small size.
- Globulins: Divided into alpha, beta, and gamma fractions based on their charge and size.
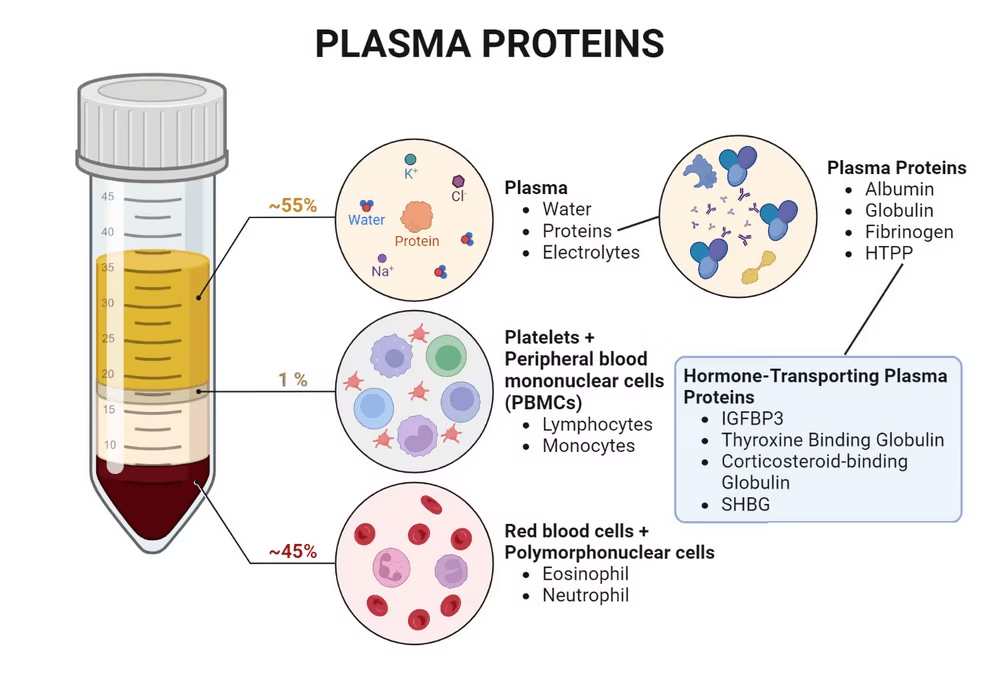
Composition
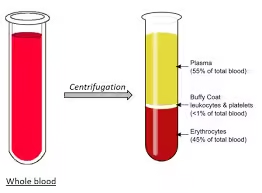
- Water (92%)
- Solids (8%)
Solids (8%)
Organic:
-
- Plasma proteins: Albumin, Globulins & Fibrinogen.
- Non-protein nitrogenous compounds: urea, free amino acids, uric acid, creatinine, creatine & NH3
- Lipids: Cholesterol, TG, phospholipids, free fatty acids
- Carbohydrates: Glucose, fructose, pentose, other substances such as Ketone bodies, bile pigments, vitamins, enzymes & hormones
Inorganic: Na+, K+, Ca2+, Mg2+, Cl–, HCO3-.
- Albumin (3.5-5.0 g/dL):
- The most abundant plasma protein (about 60% of total proteins).
- Structure: A single polypeptide chain with a high proportion of α-helices, facilitating binding with various ligands.
- Globulins (2.0-3.5 g/dL):
- Comprised of three main types:
- Alpha-1 Globulins (e.g., α1-antitrypsin): Inhibits proteolytic enzymes.
- Alpha-2 Globulins (e.g., haptoglobin): Binds free haemoglobin.
- Beta Globulins (e.g., transferrin, complement proteins): Involved in iron transport and immune functions.
- Gamma Globulins (Immunoglobulins): Essential for the immune response.
- Comprised of three main types:
- Fibrinogen (0.2-0.4 g/dL):
- A soluble glycoprotein that plays a critical role in blood coagulation.
- Other Proteins:
- Include various enzymes, hormones, and acute-phase reactants (e.g., C-reactive protein).
Functions
- Osmotic Regulation:
- Oncotic Pressure: Albumin and other proteins maintain colloid osmotic pressure, preventing fluid from leaking into tissues, which is vital for tissue hydration and blood volume maintenance.
- Transport Functions:
- Fatty Acid Transport: Albumin binds free fatty acids, allowing their transport to tissues for energy.
- Hormone Transport: Steroid hormones (e.g., cortisol) and thyroid hormones (e.g., thyroxine) are bound to specific carrier proteins.
- Metal Ion Transport: Transferrin binds iron in a soluble form for transport, ensuring iron delivery to cells.
- Immune Response:
- Antibodies: Gamma globulins (immunoglobulins) are crucial in identifying and neutralizing pathogens, including bacteria and viruses.
- Complement Proteins: Part of the immune system, these proteins help opsonize pathogens and promote inflammation.
- Clotting Mechanisms:
- Fibrinogen: Converts to fibrin in the presence of thrombin during the clotting cascade, forming a stable mesh that helps stop bleeding.
- Enzymatic and Regulatory Functions:
- Enzymes: Some plasma proteins function as enzymes, catalyzing biochemical reactions essential for metabolism.
- Regulatory Roles: Proteins like α1-antitrypsin regulate enzyme activity and protect tissues from damage.
- Buffering Capacity:
- Plasma proteins contribute to the blood’s buffering system, helping maintain pH within a narrow range, crucial for optimal enzyme function and overall metabolic balance.
- Nutrient Supply:
- Plasma proteins can serve as a reservoir of amino acids for tissue repair and synthesis, especially during stress or injury.
Serum Protein Analysis
A. Methods of Analysis
- Electrophoresis
- Overview: This technique separates proteins based on size and charge when an electric current is applied to a gel medium.
- Types:
- Agarose Gel Electrophoresis: Commonly used for serum protein electrophoresis (SPEP). Serum samples are applied to wells in an agarose gel, and the proteins migrate according to their charge and size.
- Polyacrylamide Gel Electrophoresis (PAGE): Provides higher resolution and is useful for separating smaller proteins or more complex mixtures. SDS-PAGE uses sodium dodecyl sulfate to denature proteins, ensuring they separate based on size.
- Interpretation: The resulting gel shows distinct bands. Normal serum typically displays five major protein fractions: albumin (largest band), alpha-1, alpha-2, beta, and gamma globulins. Abnormalities in these patterns can indicate specific diseases (e.g., monoclonal gammopathy in multiple myeloma).
- Immunodiffusion and Immunoelectrophoresis
- Overview: These techniques involve diffusing antibodies and antigens to form precipitin lines or bands, allowing identification and quantification.
- Usage: Particularly useful for measuring immunoglobulins and identifying specific proteins like C-reactive protein (CRP) or specific antibodies (e.g., anti-Smith antibodies in lupus).
- Interpretation: The presence and intensity of precipitin lines can indicate the concentration of specific proteins.
- Enzyme-Linked Immunosorbent Assay (ELISA)
- Overview: A sensitive technique that uses antibodies to detect and quantify proteins. It involves binding the target protein to a solid surface, followed by enzyme-linked antibodies that produce a measurable signal (usually colourimetric).
- Applications: Commonly used for hormones (e.g., insulin, cortisol), cytokines (e.g., TNF-α), and specific disease markers (e.g., HIV antibodies).
- Interpretation: The intensity of the colour change is proportional to the concentration of the target protein, allowing quantitative analysis.
- Mass Spectrometry
- Overview: A highly precise technique that analyzes proteins based on their mass-to-charge ratio. It provides detailed information on protein structure, post-translational modifications, and quantity.
- Applications: Useful for identifying proteins in complex mixtures and characterizing specific proteins, such as in biomarker discovery.
- Interpretation: Results are displayed as mass spectra, allowing the identification of protein species and modifications.
- Chromatography
- Overview: Techniques that separate proteins based on size, charge, or binding affinity. Common types include:
- Size Exclusion Chromatography: Separates proteins based on size; larger proteins elute first.
- Ion Exchange Chromatography: Separates based on charge.
- Affinity Chromatography: Uses specific interactions (e.g., with antibodies) to isolate proteins.
- Applications: Used for purifying proteins for further analysis and studying protein interactions.
- Overview: Techniques that separate proteins based on size, charge, or binding affinity. Common types include:
- Total Protein Measurement
- Overview: Determines the total concentration of proteins in serum.
- Methods:
- Biuret Method: Based on the colour change of proteins in alkaline solutions.
- Refractometry: Measures the refractive index of serum.
- Dye-Binding Assays: Use dyes (e.g., Bradford or Lowry methods) that bind to proteins, changing colour based on protein concentration.
- Interpretation: Total protein levels provide information about nutritional status, hydration, and disease presence.
B. Interpretation of Results
- Albumin Levels:
- Normal range: 3.5-5.0 g/dL.
- Decreased levels may indicate liver disease, nephrotic syndrome, malnutrition, or inflammation.
- Increased levels often occur in dehydration.
- Globulin Levels:
- Normal range: 2.0-3.5 g/dL.
- Changes in levels may suggest various conditions:
- Increased levels can indicate chronic inflammatory conditions or infections.
- Decreased levels may occur in immunodeficiencies or liver disease.
- Protein Electrophoresis Patterns:
- Normal Pattern: A smooth distribution of albumin and globulin fractions.
- Monoclonal Gammopathy: Sharp spike (M spike) in gamma globulin region, often indicative of multiple myeloma.
- Polyclonal Gammopathy: Broad-based increase in gamma globulins, typically due to chronic inflammation or infection.
- Hypoalbuminemia: Reduced albumin, often seen in liver disease or nephrotic syndrome.
Analysis of Other Body Fluids
A. Cerebrospinal Fluid (CSF)
- Collection: Obtained via lumbar puncture.
- Methods:
- Cell Count and Differential: Identifies white blood cells, indicating infection or inflammation.
- Protein Level Measurement: Normal range: 15-45 mg/dL. Elevated levels may indicate infections (e.g., meningitis), multiple sclerosis, or haemorrhage.
- Electrophoresis: Helps differentiate types of proteins present, useful for diagnosing multiple sclerosis (oligoclonal bands).
B. Synovial Fluid
- Collection: Obtained via arthrocentesis (joint aspiration).
- Methods:
- Macroscopic Examination: Assess clarity, colour, and viscosity.
- Cell Count: Differential count to identify inflammatory or infectious processes.
- Protein Concentration: Elevated levels often indicate arthritis or infection.
- Culture and Sensitivity: For infectious causes.
C. Urine
- Collection: Typically, a clean-catch midstream sample.
- Methods:
- Dipstick Test: Rapid screening for protein, glucose, ketones, etc.
- 24-Hour Urine Collection: Quantitative measurement of protein excretion (normal < 150 mg/day).
- Electrophoresis: Identifies specific protein abnormalities (e.g., Bence-Jones proteins in multiple myeloma).
D. Pleural and Peritoneal Fluid
- Collection: Obtained via thoracentesis or paracentesis.
- Methods:
- Cell Count and Differential: Assess for infection or malignancy.
- Protein Concentration: Helps differentiate between transudates and exudates using Light’s criteria (e.g., pleural fluid protein to serum protein ratio).
- Culture and Cytology: For infection or cancer.
E. Saliva
- Collection: Non-invasive; can be collected using specialized containers.
- Methods:
- Proteomic Analysis: ELISA and mass spectrometry can be used for specific biomarkers (e.g., oral cancer markers and systemic disease indicators).
- Microbial Analysis: Assess oral health and disease prevalence.
Clinical Significance
- Diagnostic Value: Serum and body fluid analyses are essential for diagnosing diseases, such as liver disorders, kidney disease, infections, and malignancies.
- Prognostic Indicators: Specific protein levels can be biomarkers for disease progression (e.g., elevated C-reactive protein in inflammatory conditions).
- Research Applications: Proteomic studies are increasingly used to discover novel biomarkers for early disease detection and monitoring.
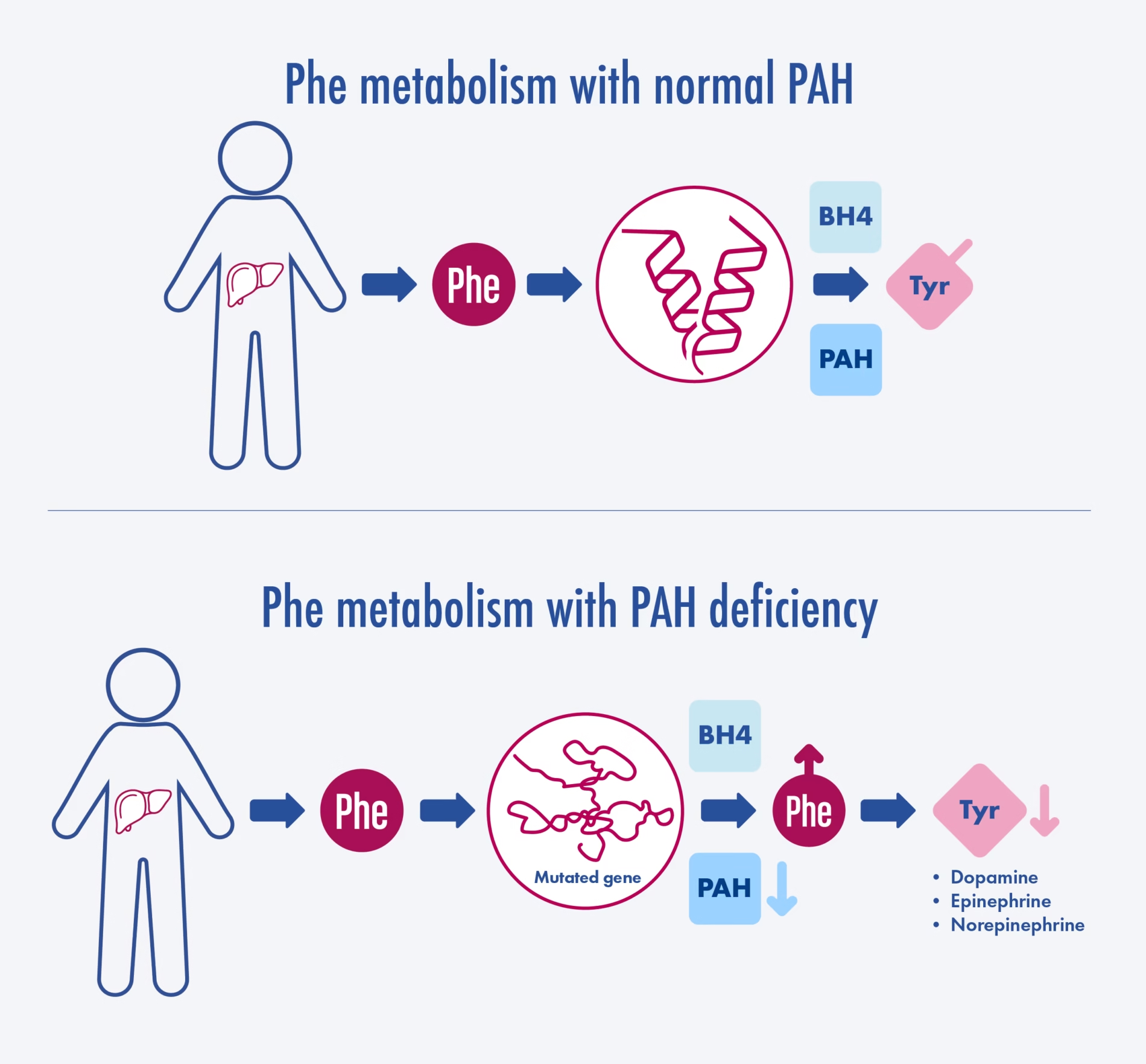
Phenylketonuria (PKU)
PKU is an autosomal recessive disorder caused by mutations in the PAH gene, leading to insufficient phenylalanine hydroxylase activity. As a result, phenylalanine accumulates in the body and is converted to phenylketones, which can be detected in the urine.
Pathophysiology
- Elevated phenylalanine levels are toxic to the brain and can cause severe neurological impairment if not managed.
- The accumulation of phenylalanine leads to secondary effects on other metabolic pathways, including impaired synthesis of neurotransmitters.
Clinical Features
Symptoms: If untreated, symptoms typically present within the first few months of life and may include:
- Intellectual disability
- Behavioural problems
- Seizures
- Hypopigmentation (due to reduced melanin production)
- Eczema
Diagnosis
- Newborn Screening: Most countries perform routine screening for PKU shortly after birth via heel prick tests that measure phenylalanine levels.
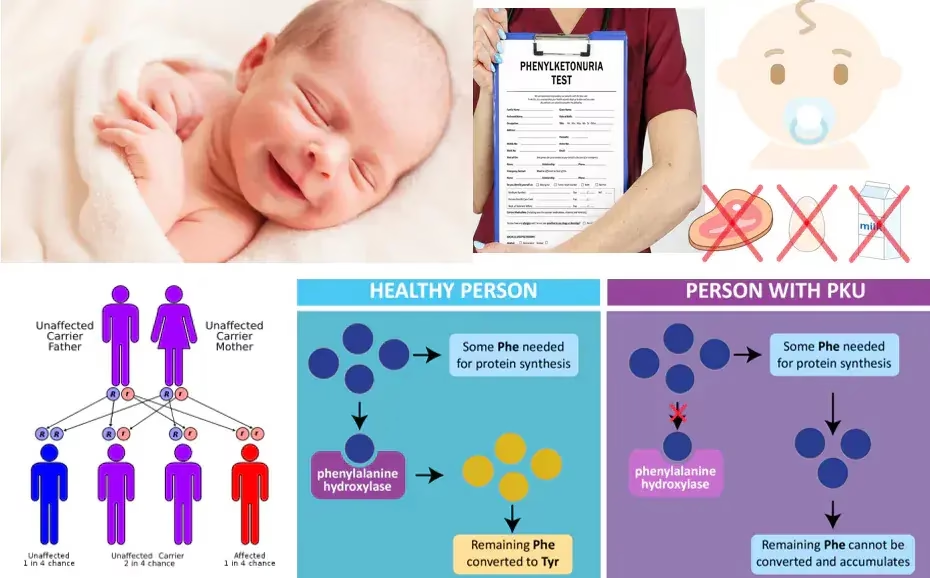
- Urine Tests: Elevated levels of phenylketones (phenylacetate, phenylpyruvate) can be detected in urine.
- Genetic Testing: Identifies mutations in the PAH gene.
Treatment
- Dietary Management: The primary treatment for PKU involves a strict diet low in phenylalanine:
-
- Patients are advised to avoid high-protein foods (e.g., meat, dairy) and certain grains.
- Special medical foods and formulas provide essential nutrients without phenylalanine.
- Supplementation: Tetrahydrobiopterin (BH4) supplementation can be effective for some patients with milder forms of PKU, enhancing residual PAH activity.
- Regular Monitoring: Patients require regular blood tests to monitor phenylalanine levels and ensure adherence to dietary restrictions.
Tyrosine disorders
Tyrosine disorders are a group of metabolic conditions associated with abnormal metabolism of the amino acid tyrosine derived from phenylalanine. These disorders can lead to various clinical manifestations and health issues. Below are the key tyrosine disorders:
1. Tyrosinemia Type I
- Cause: Caused by a deficiency of the enzyme fumarylacetoacetate hydrolase (FAH), leading to the accumulation of toxic metabolites, particularly fumarylacetoacetate and succinylacetone.
- Inheritance: Autosomal recessive.
Clinical Features
Symptoms typically appear in infancy or early childhood and may include:
- Liver failure
- Renal tubular dysfunction
- Failure to thrive
- Jaundice
- Hypoglycemia
- Neurological symptoms, including developmental delays
Diagnosis
- Newborn screening: Elevated levels of succinylacetone in urine or blood.
- Genetic Testing: Identifies mutations in the FAH gene.
Treatment
- Dietary Management: A diet low in tyrosine and phenylalanine.
- Nitisinone: A drug that inhibits the synthesis of toxic metabolites, significantly improving outcomes.
- Liver Transplantation: Considered in severe cases with liver failure.
2. Tyrosinemia Type II
- Cause: Caused by a deficiency of the enzyme tyrosine aminotransferase (TAT).
- Inheritance: Autosomal recessive.
Clinical Features
Symptoms usually present in early childhood and may include:
- Ocular symptoms (photophobia, keratitis)
- Skin lesions (erythematous and painful)
- Developmental delays
- Liver dysfunction
Diagnosis
- Urine Tests: Elevated levels of tyrosine and other metabolites.
- Blood Tests: Increased tyrosine levels.
- Genetic Testing: Confirms mutations in the TAT gene.
Treatment
- Dietary Management: Low-tyrosine diet, avoiding foods rich in tyrosine (e.g., dairy, meats).
- Symptomatic Treatment: Management of ocular and skin symptoms.
3. Tyrosinemia Type III
- Cause: Caused by the enzyme p-hydroxyphenylpyruvate dioxygenase (HPPD) deficiency.
- Inheritance: Autosomal recessive.
Clinical Features
This type is relatively rare and may present with:
- Mild developmental delays
- Ocular abnormalities
- Possible movement disorders
Diagnosis
- Urine Tests May show elevated levels of tyrosine and its metabolites.
- Blood Tests: Increased tyrosine levels.
- Genetic Testing: Confirms mutations in the HPPD gene.
Treatment
- Dietary Management: Similar to other types, focusing on reducing tyrosine intake.
- Supplementation: Some patients may benefit from vitamin C supplementation, though this is less standardized.
Alkaptonuria
Alkaptonuria is a rare autosomal recessive metabolic disorder caused by the deficiency of the enzyme homogentisate 1,2-dioxygenase (HGD), which is involved in the catabolism of tyrosine and phenylalanine.
Pathophysiology
- Defect in Metabolism:
- In normal metabolism, tyrosine is broken down into fumarate and acetoacetate. Homogentisic acid (HGA) is an intermediate in this pathway.
- In alkaptonuria, the HGD enzyme is deficient, leading to the accumulation of homogentisic acid in the body.
- Accumulation of Homogentisic Acid:
- When exposed to air, HGA is excreted in the urine and can oxidize to form a dark pigment.
- This pigment can also deposit in connective tissues, leading to a condition known as ochronosis, where tissues darken and become damaged over time.
Clinical Features
- Urinary Changes:
- Darkening of urine upon exposure to air due to the oxidation of HGA.
- Musculoskeletal Symptoms:
- Early onset of arthritis, especially in large joints (e.g., hips, knees).
- Joint pain and stiffness can develop in middle adulthood.
- Connective Tissue Changes:
- Blue-black discolouration of the sclera and other tissues due to pigment deposition.
- Calcification of cartilage (especially in the spine and joints).
- Other Symptoms:
- Possible cardiovascular issues due to pigment deposits in heart valves.
- Kidney stones may occur due to HGA crystallization.
Enzyme Defects
- Deficiency:
- The primary defect is in the enzyme homogentisate 1,2-dioxygenase (HGD).
- This enzyme catalyzes the conversion of homogentisic acid to maleylacetoacetate in the normal catabolism of tyrosine.
- Genetics:
- Caused by mutations in the HGD gene, located on chromosome 3 (3q21.1).
Biochemical Manifestations
- Urinary Findings:
- Elevated levels of homogentisic acid in urine (can be detected via urine tests).
- Urine turns dark when exposed to air due to oxidation.
- Plasma Findings:
- Elevated levels of tyrosine and phenylalanine can be observed due to their impaired metabolism.
- Tissue Deposits:
- HGA accumulates in connective tissues, leading to pigmentation and damage.
Diagnosis
- Urine Test:
- Qualitative Test: Urine can be subjected to a simple test where it turns dark upon exposure to air.
- Quantitative Test: High-performance liquid chromatography (HPLC) can measure levels of homogentisic acid.
- Genetic Testing:
- Identification of mutations in the HGD gene can confirm diagnosis.
- Clinical Evaluation:
- Assessment of symptoms, family history, and physical examination to identify typical features of alkaptonuria.
Treatment
- Management:
- Symptomatic Treatment: Pain relief and management of arthritis symptoms with non-steroidal anti-inflammatory drugs (NSAIDs).
- Physical Therapy: To improve joint function and mobility.
- Dietary Restrictions:
- While there are no strict dietary guidelines, limiting tyrosine and phenylalanine intake may be beneficial, although not always necessary.
- Surgical Intervention:
- Joint replacement surgery may be required in cases of severe arthritis or joint damage.
- Research:
- There are ongoing studies into the use of nitisinone, a drug used in tyrosinemia, to potentially lower levels of homogentisic acid in the body.
Melanuria
Melanuria is a condition characterized by melanin or its metabolites in the urine. It is typically associated with disorders that affect melanin production or metabolism, particularly conditions related to melanoma, a type of skin cancer.
Causes
- Melanoma:
- Primary Cutaneous Melanoma: The most common cause; malignant transformation of melanocytes can lead to excess melanin production.
- Metastatic Melanoma: Advanced stages can lead to the release of melanin into the bloodstream.
- Other Conditions:
- Nevi (Moles): Changes in benign pigmented lesions can sometimes lead to melanuria.
- Pigmented Nephropathy: Conditions that cause renal damage and lead to the release of melanin into the urine.
- Certain Inborn Errors of Metabolism: Rare metabolic disorders may disrupt normal melanin metabolism.
Pathophysiology
- In cases of melanoma, malignant cells may produce excessive amounts of melanin. When these cells die or are damaged, melanin can be released into the bloodstream and filtered by the kidneys, resulting in melanuria.
- Other pathways may involve the conversion of tyrosine (the precursor of melanin) into melanin and its metabolites, which may also be excreted in urine.
Clinical Features
- Urinary Changes: The urine may appear dark brown or black due to melanin.
- Associated Symptoms: Patients may have symptoms related to the underlying cause, such as:
- Skin lesions or changes (in melanoma).
- Signs of metastasis (e.g., bone pain, organ dysfunction).
- Other symptoms related to kidney function if renal involvement is present.
Diagnosis
- Urinalysis:
- A standard urinalysis can indicate the presence of dark urine, and specific tests can confirm the presence of melanin or its metabolites.
- Clinical History:
- Evaluation of the patient’s history, including any known skin lesions, prior diagnoses of melanoma, or other related conditions.
- Imaging and Biopsy:
- Imaging studies (e.g., ultrasound, CT scan) to assess for tumours or metastases.
- Skin biopsy of suspicious lesions to confirm melanoma.
- Biochemical Tests:
- Measuring urinary metabolites associated with melanin production, such as 5-hydroxyindoleacetic acid (5-HIAA) or other relevant markers.
Treatment
- Management of Underlying Cause: The treatment of melanuria is primarily focused on addressing the underlying condition, especially if it is due to melanoma.
- Surgical Intervention: Removal of melanoma or metastases may be necessary.
- Chemotherapy and Immunotherapy: Standard treatments for advanced melanoma to reduce tumour burden and associated symptoms.
- Supportive Care: Management of melanoma-related symptoms or any renal implications.
Cystinuria
Cystinuria is a genetic disorder characterized by the impaired reabsorption of certain amino acids, particularly cysteine, in the kidneys. This leads to high levels of cystine and other dibasic amino acids (ornithine, lysine, and arginine) in the urine, forming kidney stones.
Pathophysiology
- Genetic Basis:
- Cystinuria is primarily caused by mutations in the SLC7A9 and SLC3A1 genes, which encode transporters responsible for the reabsorption of cystine and other dibasic amino acids in the renal proximal tubules.
- These mutations lead to defective cystine transport, causing it to accumulate in the urine.
- Formation of Cystine Stones:
- Cystine is poorly soluble in urine. When its concentration exceeds a certain threshold (usually around 250 mg/dL), it can precipitate, forming cystine crystals and stones in the kidneys and urinary tract.
Clinical Features
- Renal Stones:
- Patients may present with symptoms related to kidney stones, such as:
- Flank pain (renal colic)
- Hematuria (blood in urine)
- Urinary obstruction
- Frequent urinary infections
- Patients may present with symptoms related to kidney stones, such as:
- Age of Onset:
- Symptoms often begin in childhood or early adulthood.
- Other Symptoms:
- Some patients may experience growth retardation due to amino acid imbalances, but many are asymptomatic until stone formation occurs.
Diagnosis
- Urine Analysis:
- 24-hour Urine Collection: Elevated levels of cystine, lysine, ornithine, and arginine in urine.
- Cystine Crystals: Urinalysis may reveal hexagonal crystals under polarized light.
- Genetic Testing:
- Identifies mutations in the SLC7A9 or SLC3A1 genes, confirming the diagnosis.
- Imaging Studies:
- Ultrasound or CT Scan: To identify the presence and size of kidney stones.
Treatment
- Hydration:
- Increasing fluid intake is critical to dilute urine and reduce cystine concentration, helping prevent stone formation.
- Dietary Modifications:
- Reducing sodium intake may help decrease cystine excretion.
- Limiting protein intake can also be beneficial.
- Medications:
- Cystine-Lowering Agents:
- Alpha-mercaptopropionylglycine (MPG): A cystine-lowering agent that can help reduce urinary cystine levels.
- Penicillamine: An alternative medication that can help solubilize cystine.
- Potassium Citrate: May be prescribed to alkalinize urine, further promoting cystine solubility.
- Cystine-Lowering Agents:
- Surgical Intervention:
- Surgical removal of stones may be necessary in large stones or recurrent urinary obstruction cases.
Homocystinuria
Homocystinuria is a rare genetic disorder characterized by the accumulation of homocysteine in the blood and urine due to defects in the metabolism of the amino acid methionine. It is primarily associated with gene mutations responsible for enzymes involved in the transsulfuration pathway.
Pathophysiology
- Genetic Basis:
- It is most commonly caused by a deficiency of the enzyme cystathionine beta-synthase (CBS), which converts homocysteine to cystathionine.
- Other rare forms, such as methylenetetrahydrofolate reductase (MTHFR) or cystathionine gamma-lyase (CGL), may involve enzyme deficiencies.
- Homocysteine Accumulation:
- With impaired conversion of homocysteine, it accumulates in the blood (hyperhomocysteinemia) and is excreted in the urine (homocystinuria).
- Elevated homocysteine levels can lead to various complications, including cardiovascular issues and connective tissue abnormalities.
Clinical Features
- Ocular Symptoms:
- Lens Dislocation: Ectopia lentis is common, leading to visual problems.
- Myopia: Increased risk of nearsightedness.
- Skeletal Features:
- Marfanoid Habitus: Patients may exhibit long limbs and arachnodactyly (long fingers).
- Skeletal Deformities: Scoliosis, osteoporosis, and other bone abnormalities may occur.
- Neurological Symptoms:
- Developmental delays and intellectual disability may be observed.
- Increased risk of seizures and psychiatric disorders.
- Vascular Complications:
- Increased risk of thromboembolic events, such as deep vein thrombosis or stroke, due to the damaging effects of elevated homocysteine on blood vessels.
Diagnosis
- Biochemical Testing:
- Plasma Homocysteine Levels: Elevated levels are indicative of homocystinuria.
- Urine Analysis: Homocysteine is present in the urine; it can be detected with specific tests.
- Genetic Testing:
- Identifies mutations in the CBS gene or other relevant genes involved in homocysteine metabolism.
- Newborn Screening:
- Some regions include homocystinuria in routine newborn screening and identifying affected infants early for prompt management.
Treatment
- Dietary Management:
- Methionine Restriction: Reducing dietary intake of methionine (found in high-protein foods) can help lower homocysteine levels.
- Supplementation:
- Vitamin B6 (Pyridoxine): Some patients respond well to high doses of vitamin B6, which can help improve CBS enzyme activity.
- Vitamin B12 and Folate: These vitamins may also help lower homocysteine levels and should be included in management.
- Medications:
- Other medications (like betaine) can help reduce homocysteine levels by promoting its conversion to other metabolites in cases where dietary and vitamin interventions are insufficient.
- Monitoring:
- Regular follow-up with blood tests to monitor homocysteine levels and adjust treatment accordingly.
Cystinosis
Cystinosis is a rare, inherited metabolic disorder characterized by the accumulation of cystine, an amino acid, within lysosomes due to a defect in the cystine transporter. This condition primarily affects the kidneys but can also have systemic effects.
Pathophysiology
- Genetic Basis:
- Cystinosis is caused by mutations in the CTNS gene, which encodes the cystine transporter protein. This transporter is responsible for moving cystine out of lysosomes.
- The defective transporter leads to the accumulation of cystine crystals in lysosomes, causing cellular damage and dysfunction.
- Accumulation of Cystine:
- High cystine levels can result in lysosomal dysfunction and damage to various organs, particularly the kidneys, eyes, and other tissues.
Clinical Features
- Renal Symptoms:
- Fanconi Syndrome: A condition characterized by:
- Renal tubular dysfunction leads to the loss of bicarbonate, phosphate, glucose, and amino acids in the urine.
- Symptoms such as growth failure, dehydration, and rickets in children.
- Progressive Renal Failure: Often leads to end-stage renal disease in adolescence or early adulthood.
- Fanconi Syndrome: A condition characterized by:
- Ocular Symptoms:
- Corneal Deposits: Cystine crystals accumulate in the cornea, leading to photophobia, corneal opacity, and potential vision loss.
- Eye Symptoms: Patients may experience burning, tearing, and sensitivity to light.
- Growth and Development:
- Delayed growth and pubertal development due to renal dysfunction and nutrient loss.
- Other Symptoms:
- Muscle weakness and pain.
- Potential endocrine dysfunction due to renal impairment.
Diagnosis
- Clinical Evaluation:
- Assessment of symptoms, family history, and physical examination.
- Urine Analysis:
- Cystine Levels: Measurement of cystine in urine; elevated levels indicate cystinosis.
- Fanconi Syndrome Indicators: Presence of glucose, phosphate, and amino acids in urine.
- Genetic Testing:
- Identifies mutations in the CTNS gene to confirm the diagnosis.
- Corneal Examination:
- A slit-lamp examination can reveal corneal deposits of cystine.
Treatment
- Cystine-Lowering Therapy:
- Cysteamine: An oral medication that reduces cystine levels by converting cystine to cysteine and facilitating its exit from lysosomes. This is the primary treatment for cystinosis.
- Cysteamine Eye Drops: To manage ocular symptoms and reduce corneal deposits.
- Supportive Care:
- Renal Management: Monitoring and managing renal function, including treatment for Fanconi syndrome.
- Dietary Modifications: Ensure adequate intake of nutrients and manage electrolyte imbalances.
- Renal Replacement Therapy:
- In cases of advanced renal failure, patients may require dialysis or kidney transplantation.
- Regular Monitoring:
- Lifelong follow-up with a multidisciplinary team to monitor kidney function, ocular health, and overall development.
Organic Acidurias
- Organic acidurias are a group of inherited metabolic disorders characterized by the accumulation of organic acids in the body due to deficiencies in specific enzymes involved in amino acid or fatty acid metabolism.
- These disorders often lead to metabolic acidosis and a variety of clinical symptoms.
Pathophysiology
- Enzyme Deficiencies:
- Organic acidurias result from defects in enzymes responsible for metabolizing organic acids. This leads to the accumulation of organic acids in the blood and urine.
- Common enzyme deficiencies include:
- Propionyl-CoA carboxylase (Propionic aciduria)
- Methylmalonyl-CoA mutase (Methylmalonic aciduria)
- Isovaleryl-CoA dehydrogenase (Isovaleric aciduria)
- 3-hydroxy-3-methylglutaryl-CoA lyase (3-Hydroxy-3-methylglutaryl-CoA aciduria)
- Metabolic Pathways:
- These enzymes involve various metabolic pathways, including amino acid catabolism and fatty acid oxidation. Deficiency leads to toxic accumulation and disruption of normal metabolic processes.
Clinical Features
- Acute Symptoms:
- Symptoms often present in infancy or early childhood and may include:
- Vomiting
- Poor feeding
- Lethargy
- Hypotonia (decreased muscle tone)
- Seizures
- Developmental delays
- Symptoms often present in infancy or early childhood and may include:
- Chronic Symptoms:
- If untreated, children may experience recurrent metabolic crises, growth failure, and neurological impairment.
- Specific Symptoms by Type:
- Propionic Aciduria: Ketoacidosis, metabolic acidosis, neurological symptoms.
- Methylmalonic Aciduria: Similar to propionic aciduria, with possible developmental delays.
- Isovaleric Aciduria: Characterized by a distinctive odour of sweaty feet due to isovaleric acid.
- 3-Hydroxy-3-Methylglutaryl-CoA Aciduria: This may lead to metabolic crises and neurological symptoms.
Diagnosis
- Urine Analysis:
- Organic Acid Profile: Urine can be tested for specific organic acids, which helps identify the type of organic aciduria.
- Gas Chromatography-Mass Spectrometry (GC-MS): A sensitive technique to detect and quantify organic acids in urine.
- Blood Tests:
- Metabolic Screening: Elevated levels of specific organic acids in the blood can indicate a disorder.
- Acidosis: Metabolic acidosis may be detected through arterial blood gas analysis.
- Genetic Testing:
- Determining mutations in specific genes associated with organic acidurias can confirm the diagnosis.
Treatment
- Dietary Management:
- Protein Restriction: Limiting dietary intake of proteins that contribute to organic acid production.
- Specific Dietary Adjustments: Patients with methylmalonic aciduria may need to avoid certain amino acids.
- Supplementation:
- Co-factor Supplementation: In some cases, supplements like vitamin B12 (for methylmalonic aciduria) may be beneficial.
- Carnitine Supplementation: May help in certain organic acidurias to improve fatty acid oxidation and reduce toxic metabolites.
- Management of Acute Crises:
- Hospitalization: Patients experiencing metabolic crises may require intravenous fluids, correction of acidosis, and management of electrolytes.
- Long-term Monitoring:
- Regular follow-up to monitor growth, development, and metabolic status is essential.