Introduction
-
Lipid metabolism refers to the processes involved in the digestion, absorption, synthesis, transport, and breakdown of lipids in the body.
-
Lipids are an important source of energy, providing about 9 kcal of energy per gram, which is higher than carbohydrates and proteins.
-
The major dietary lipids include triglycerides, phospholipids, cholesterol, and fatty acids, which are essential for various biological functions.
-
Lipids play several important roles in the body such as energy storage, formation of cell membranes, hormone synthesis, and insulation of organs.
-
The metabolism of lipids mainly occurs in organs such as the liver, adipose tissue, skeletal muscles, and intestines.
-
Lipid metabolism involves several metabolic pathways including lipolysis, β-oxidation of fatty acids, ketone body formation, and lipid biosynthesis.
-
Hormones such as insulin, glucagon, and epinephrine regulate lipid metabolism by controlling the breakdown and synthesis of fats.
-
Proper lipid metabolism is essential for maintaining energy balance, cellular structure, and overall metabolic health, while disturbances may lead to diseases such as obesity, fatty liver, and cardiovascular disorders.
- Metabolism of Lipids includes a wide variety of chemical substances
Such as:- Neutral fat (triacylglycerol or triglycerides)
- Fatty acids and their derivatives
- Phospholipids
- Glycolipids
Fatty acid oxidation
Fatty acid oxidation, or β-oxidation, is the metabolic process through which fatty acids are broken down to generate energy. This process primarily occurs in the mitochondria of cells and involves three steps.
Activation of Fatty Acids
- Process:
- Fatty acids are activated in the cytoplasm before being transported into the mitochondria.
- Acyl-CoA Synthetase (Fatty Acid Thiokinase) catalyzes the reaction.
- Reaction Mechanism:
- The reaction consists of two key steps:
-
- Formation of Acyl-AMP:
- The fatty acid reacts with ATP, releasing pyrophosphate (PPi) and forming acyl-adenylate (acyl-AMP).
- Formation of Fatty Acyl-CoA:
- Coenzyme A (CoA) attacks the acyl-AMP, releasing AMP and forming acyl-CoA.
- Formation of Acyl-AMP:
- Energetics:
- The hydrolysis of PPi (to two inorganic phosphates) is energetically favourable and drives the reaction forward. The process effectively consumes one ATP molecule, converting it to AMP and pyrophosphate.
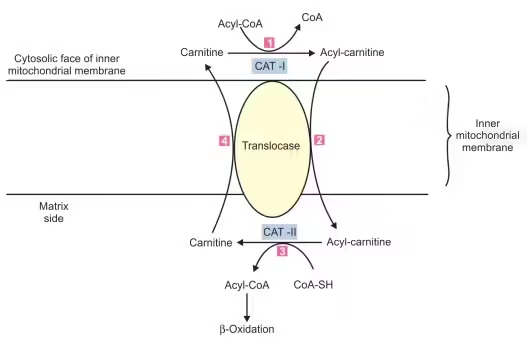
Transport into Mitochondria
- Carnitine Shuttle:
- The carnitine shuttle facilitates the transport of long-chain fatty acids into the mitochondria, crucial for maintaining metabolic efficiency.
- Steps:
-
- Conversion to Acylcarnitine:
- Carnitine Acyltransferase I (CPT I) catalyzes the reaction of fatty acyl-CoA with carnitine to form acylcarnitine.
- Translocation:
- Acylcarnitine is shuttled into the mitochondrial matrix via a specific translocase.
- Conversion back to Acyl-CoA:
- Once inside, Carnitine Acyltransferase II (CPT II) converts acylcarnitine back to fatty acyl-CoA, regenerating carnitine for the shuttle.
- Conversion to Acylcarnitine:
- Clinical Implications:
-
- Deficiencies in carnitine or enzymes involved in the shuttle can lead to fatty acid oxidation disorders, causing hypoglycemia and muscle weakness during fasting.
β-Oxidation in Mitochondria
Once inside the mitochondria, β-oxidation proceeds through a series of four enzymatic reactions that remove two-carbon units from the fatty acyl-CoA: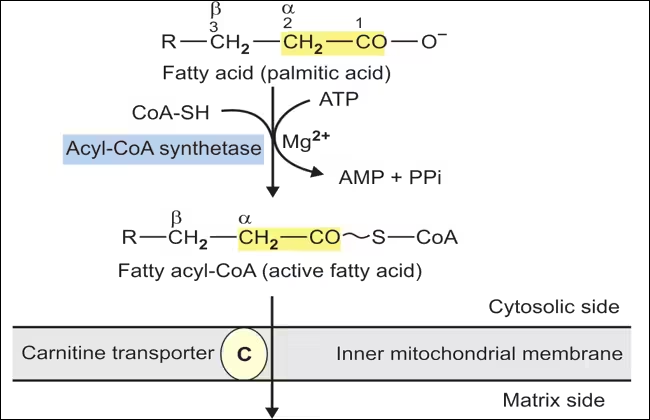
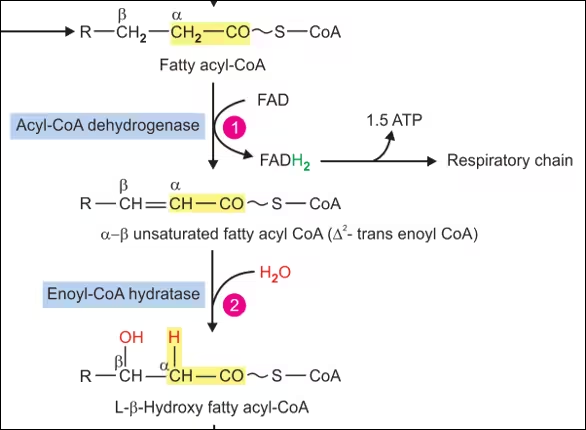
- Dehydrogenation:
- Enzyme: Acyl-CoA Dehydrogenase.
- Reaction: Fatty acyl-CoA is oxidized, and a double bond is introduced between the α and β carbons, forming trans-Δ²-enoyl-CoA.
- Co-factors: FAD is reduced to FADH₂, contributing to the electron transport chain for ATP production.
- Hydration:
- Enzyme: Enoyl-CoA Hydratase.
- Reaction: Water is added to trans-Δ²-enoyl-CoA, converting it into L-3-hydroxyacyl-CoA.
- Significance: This step is important for positioning the hydroxyl group for the subsequent oxidation.
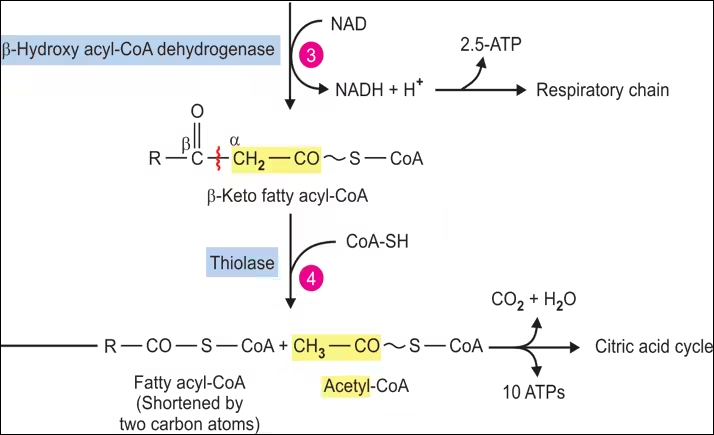
- Dehydrogenation:
- Enzyme: 3-Hydroxyacyl-CoA Dehydrogenase.
- Reaction: L-3-hydroxyacyl-CoA is oxidized to 3-ketoacyl-CoA, with NAD⁺ reduced to NADH.
- Co-factors: NADH enters the electron transport chain for ATP production.
- Thiolysis:
- Enzyme: β-Ketothiolase.
- Reaction: 3-ketoacyl-CoA is cleaved by CoA, releasing a shorter fatty acyl-CoA (two carbons shorter) and acetyl-CoA.
- Product: Acetyl-CoA can enter the citric acid cycle.
- Cycle Continuation:
- The process repeats, with the shortened fatty acyl-CoA re-entering β-oxidation until the entire fatty acid chain is converted into acetyl-CoA units.
End Products and Energy Yield
- For each cycle of β-oxidation, the following are produced:
- 1 Acetyl-CoA: Enters the citric acid cycle for energy production.
- 1 FADH₂: Produces approximately 1.5 ATP via oxidative phosphorylation.
- 1 NADH: Produces approximately 2.5 ATP via oxidative phosphorylation.
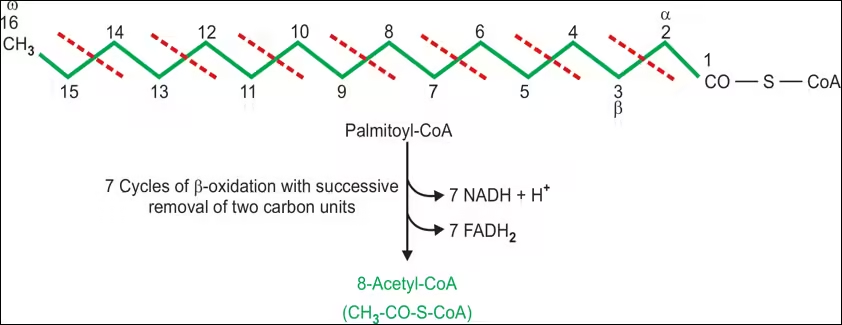
- Example: Complete oxidation of palmitic acid (C16:0):
- Total yield:
- 8 Acetyl-CoA: Each enters the citric acid cycle.
- 7 NADH: Total ATP from 7 NADH = 7 × 2.5 = 17.5 ATP.
- 7 FADH₂: Total ATP from 7 FADH₂ = 7 × 1.5 = 10.5 ATP.
- Total ATP from β-oxidation = 17.5 + 10.5 = 28 ATP.
- From Citric Acid Cycle: Each Acetyl-CoA generates approximately 10 ATP (3 NADH, 1 FADH₂, 1 GTP/ATP), leading to an additional 80 ATP from the 8 Acetyl-CoA.
- Overall Total ATP Yield: ~108 ATP from palmitic acid (considering 2 ATP consumed for activation).
- Total yield:
Regulation of Fatty Acid Oxidation
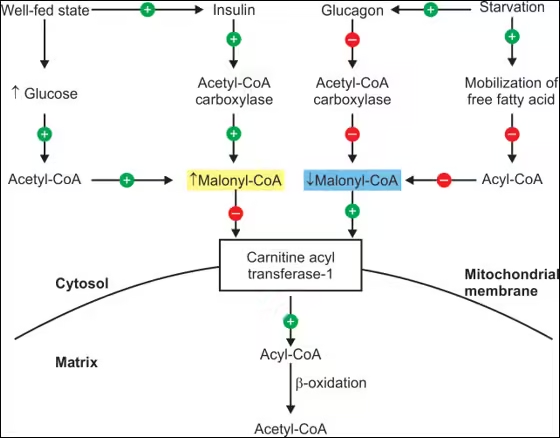
- Hormonal Regulation:
- Insulin: Inhibits fatty acid oxidation by promoting fat storage and synthesis.
- Glucagon and Epinephrine: Stimulate lipolysis in adipose tissue, increasing free fatty acids in the bloodstream available for oxidation.
- Nutritional Status:
- Increased fatty acid oxidation occurs during fasting, prolonged exercise, or low-carbohydrate diets, where the body relies more on fat stores for energy.
- Substrate Availability:
- High levels of malonyl-CoA (an intermediate in fatty acid synthesis) inhibit CPT I, reducing fatty acid entry into mitochondria. Conversely, low malonyl-CoA levels favour fatty acid oxidation.
- Mitochondrial Function:
- Proper mitochondrial function and integrity are crucial for effective β-oxidation and ATP production.
Alternate Pathways and Considerations
- Peroxisomal β-Oxidation:
- Occurs in peroxisomes for very long-chain fatty acids (C20:0 and above). This process is similar to mitochondrial β-oxidation but produces hydrogen peroxide and shorter-chain fatty acids.
- Enzymatic steps and transport mechanisms differ slightly, and acyl-CoA oxidase catalyzes the initial dehydrogenation.
- Ketogenesis:
- It occurs when there is an excess of acetyl-CoA, particularly during fasting or uncontrolled diabetes.
- Acetyl-CoA is converted to ketone bodies (acetoacetate, β-hydroxybutyrate, and acetone) in the liver, which can be used as an alternative energy source by various tissues, including the brain.
Clinical Implications
- Fatty Acid Oxidation Disorders:
- Genetic defects in enzymes involved in fatty acid oxidation (e.g., CPT I, CPT II, and acyl-CoA dehydrogenases) can lead to severe metabolic disorders.
- Symptoms include hypoglycemia, muscle weakness, and, in severe cases, sudden death due to energy depletion.
- Impact of Diet and Lifestyle:
- Diets high in carbohydrates may reduce fatty acid oxidation, leading to fat accumulation and obesity.
- Regular physical activity can enhance fatty acid oxidation and improve metabolic health.
- Zellweger syndrome
- Refsum’s disease
Metabolism of Ketone Bodies
- Ketone bodies are important metabolic molecules produced primarily in the liver during prolonged fasting, carbohydrate restriction, or increased fatty acid oxidation.
- They serve as an alternative energy source for tissues, especially the brain when glucose availability is low.
Types of Ketone Bodies
There are three main types of ketone bodies:
- Acetoacetate:
-
- The first ketone body is produced during ketogenesis.
- It can be converted into the other two ketone bodies or used directly for energy.
- β-Hydroxybutyrate:
-
- Formed from acetoacetate in a reduction reaction that requires NADH.
- While technically not a true ketone (due to its alcohol functional group), it is often grouped with ketone bodies because of its role as an energy substrate.
- Acetone:
-
- A minor ketone body is produced in smaller amounts.
- It is generated from the spontaneous decarboxylation of acetoacetate and is volatile, leading to its excretion via breath.
Production of Ketone Bodies (Ketogenesis)
- Ketogenesis occurs primarily in the liver, particularly in the mitochondria of hepatocytes.
- The process is initiated when there is an increased availability of acetyl-CoA, typically from fatty acid oxidation.
Steps of Ketogenesis: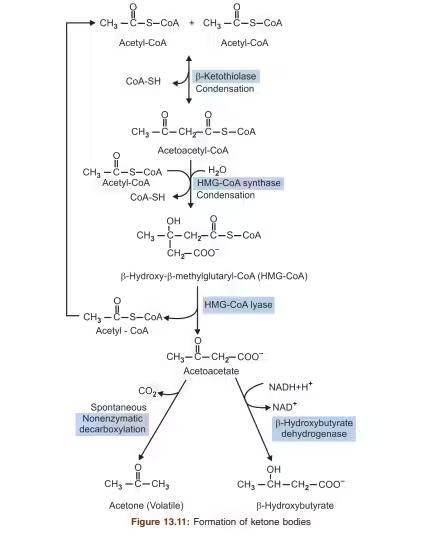
- Formation of Acetoacetyl-CoA:
- Two molecules of acetyl-CoA combine to form acetoacetyl-CoA.
- Enzyme: Thiolase.
- Formation of HMG-CoA:
- Acetoacetyl-CoA is converted to HMG-CoA (3-hydroxy-3-methylglutaryl-CoA).
- Enzyme: HMG-CoA Synthase.
- Production of Acetoacetate:
- HMG-CoA is then cleaved to produce acetoacetate and acetyl-CoA.
- Enzyme: HMG-CoA Lyase.
- Conversion to β-Hydroxybutyrate:
- Acetoacetate can be reduced to β-hydroxybutyrate using NADH.
- Enzyme: β-Hydroxybutyrate Dehydrogenase.
- Formation of Acetone:
- Acetoacetate can spontaneously decarboxylate to form acetone, which is then exhaled.
Utilization of Ketone Bodies
Ketone bodies are transported from the liver to other tissues (like the brain, heart, and muscles) where they are used as an energy source.
Steps for Utilization:
- Transport into Cells:
- Ketone bodies are transported into cells via monocarboxylate transporters (MCTs).
- Conversion back to Acetyl-CoA:
- Inside the cells, β-hydroxybutyrate is converted back to acetoacetate and then to acetoacetyl-CoA.
- Enzyme: β-Ketoacyl-CoA Transferase (only present in peripheral tissues, not in the liver).
- Entry into the Citric Acid Cycle:
- Acetoacetyl-CoA can be split into two molecules of acetyl-CoA, which then enter the citric acid cycle to generate ATP.
Physiological Functions
- Energy Source: Ketone bodies provide an efficient energy source, particularly during fasting, prolonged exercise, or carbohydrate-restricted diets.
- Brain Fuel: The brain can derive up to 75% of its energy from ketone bodies during prolonged fasting or low-carbohydrate intake, reducing its reliance on glucose.
- Sparing Glucose: By utilizing ketone bodies, the body conserves glucose for tissues that require it, like red blood cells and certain brain functions.
Clinical Significance
- Diabetes Mellitus: In uncontrolled diabetes (particularly type 1), insufficient insulin leads to increased fatty acid mobilization and ketogenesis, potentially resulting in diabetic ketoacidosis (DKA), a life-threatening condition characterized by high levels of ketone bodies in the blood and urine.
- Starvation: Prolonged fasting leads to elevated ketone body levels as the body shifts to fat oxidation for energy.
- Low-Carbohydrate Diets: Ketogenic diets are designed to induce ketosis for weight loss and management of certain medical conditions, such as epilepsy.
- Ketoacidosis
- Ketosis
Measurement:
- Ketonuria: The presence of ketone bodies in urine indicates increased ketone production, often seen in uncontrolled diabetes or fasting.
- Ketoacidosis: High levels of ketones can lead to metabolic acidosis, which is clinically significant in conditions like DKA.
Regulation of Ketogenesis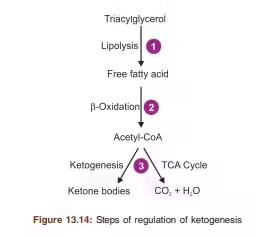
- Hormonal Control: Insulin levels play a crucial role in regulating ketogenesis.
-
- Low Insulin: Stimulates lipolysis and ketogenesis.
- High Insulin: Inhibits ketogenesis by promoting glucose utilization and fatty acid synthesis.
- Substrate Availability: High levels of fatty acids and low levels of carbohydrates promote ketone body production.
Metabolism of Cholesterol
- Cholesterol metabolism is a vital biochemical process involving the synthesis, transport, regulation, and degradation of cholesterol, a crucial lipid molecule in the body.
- Cholesterol is essential in cell membrane structure, hormone production, and bile acid synthesis.
- Here’s a detailed overview of cholesterol metabolism, including its pathways, regulation, and clinical significance.
Structure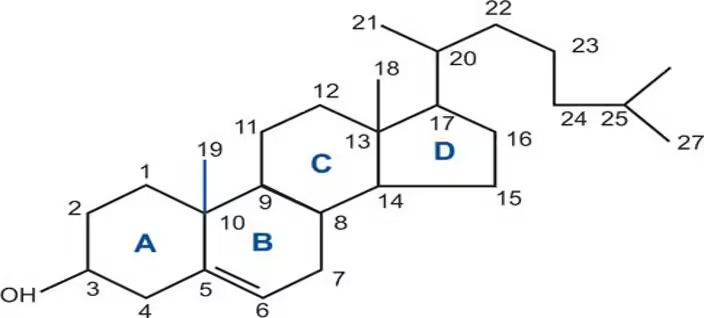
Biosynthesis of Cholesterol
Cholesterol biosynthesis is a multi-step enzymatic process that primarily occurs in the liver but can also happen in other tissues. Here’s a detailed breakdown:
A. Stages of Cholesterol Biosynthesis
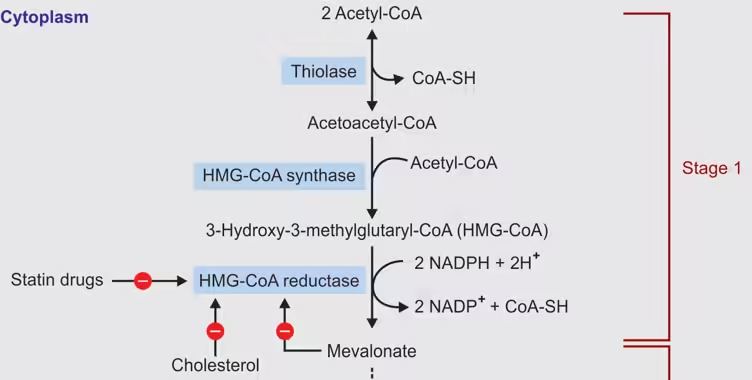

- Formation of Acetyl-CoA:
- Glycolysis: Glucose is metabolized to pyruvate, which is converted to acetyl-CoA in the mitochondria by the enzyme pyruvate dehydrogenase.
- Fatty Acid Oxidation: Fatty acids are broken down into acetyl-CoA units.
- Amino Acid Catabolism: Some amino acids can also be converted to acetyl-CoA.
- Synthesis of HMG-CoA:
- Enzyme: HMG-CoA synthase catalyzes the condensation of three acetyl-CoA molecules to form HMG-CoA.
- Reduction to Mevalonate:
- Key Enzyme: HMG-CoA reductase catalyzes the reduction of HMG-CoA to mevalonate. This is the rate-limiting step in cholesterol synthesis.
- Regulation: Statins inhibit this enzyme, lowering cholesterol levels.
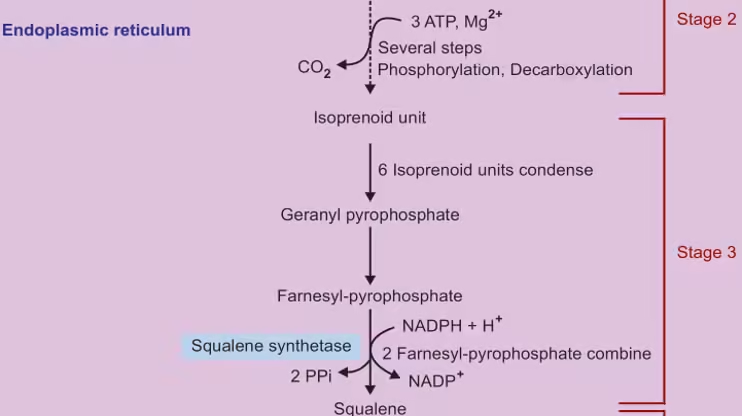
- Conversion of Mevalonate to Isoprenoid Units:
- Mevalonate undergoes phosphorylation by mevalonate kinase and phosphomevalonate kinase to form 5-pyrophosphomevalonate.
- Decarboxylation by mevalonate decarboxylase produces isopentenyl pyrophosphate (IPP).
- Formation of Squalene:
- IPP is converted to farnesyl pyrophosphate (FPP) through condensation reactions.
- Two FPP molecules condense to form squalene, catalyzed by squalene synthase.
- Cyclization of Squalene:
- Squalene undergoes a series of enzymatic reactions, including cyclization, to form lanosterol.
- Lanosterol is then converted to cholesterol through several steps involving demethylation and isomerization.
B. Cholesterol Structure and Function
- Cholesterol is a sterol with a four-ring hydrocarbon structure, making it essential for:
- Cell Membrane Integrity: It maintains membrane fluidity and structural integrity.
- Hormone Synthesis: Precursor for steroid hormones (e.g., cortisol, estrogen, testosterone).
- Vitamin D Synthesis: Precursor for vitamin D, crucial for calcium metabolism.
Degradation of Cholesterol
Cholesterol is primarily degraded through conversion to bile acids, facilitating lipid digestion and absorption.
A. Bile Acid Biosynthesis
- Conversion to Bile Acids:
- Cholesterol is converted to bile acids through a series of oxidative reactions, predominantly in the liver.
- Key Enzymes:
- CYP7A1: Converts cholesterol to 7α-hydroxycholesterol, the first step in bile acid synthesis.
- Other cytochrome P450 enzymes modify this intermediate to produce primary bile acids (cholic acid and chenodeoxycholic acid).
- Conjugation:
- Primary bile acids are conjugated with glycine or taurine to form bile salts, increasing their solubility.
- This process is catalyzed by bile acid-CoA acid N-acyltransferase.
- Storage and Secretion:
- Bile salts are stored in the gallbladder and released into the intestines to emulsify dietary fats, aiding digestion and absorption.
B. Cholesterol Excretion
- Bile: Cholesterol can be directly excreted into the bile. The liver regulates the amount of cholesterol excreted in bile to maintain homeostasis.
- Faecal Excretion: Some cholesterol is excreted in faeces, especially after dietary intake.
Cholesterol Transport Mechanisms
Cholesterol is transported in the blood as part of lipoproteins. Each type of lipoprotein plays a distinct role in cholesterol metabolism: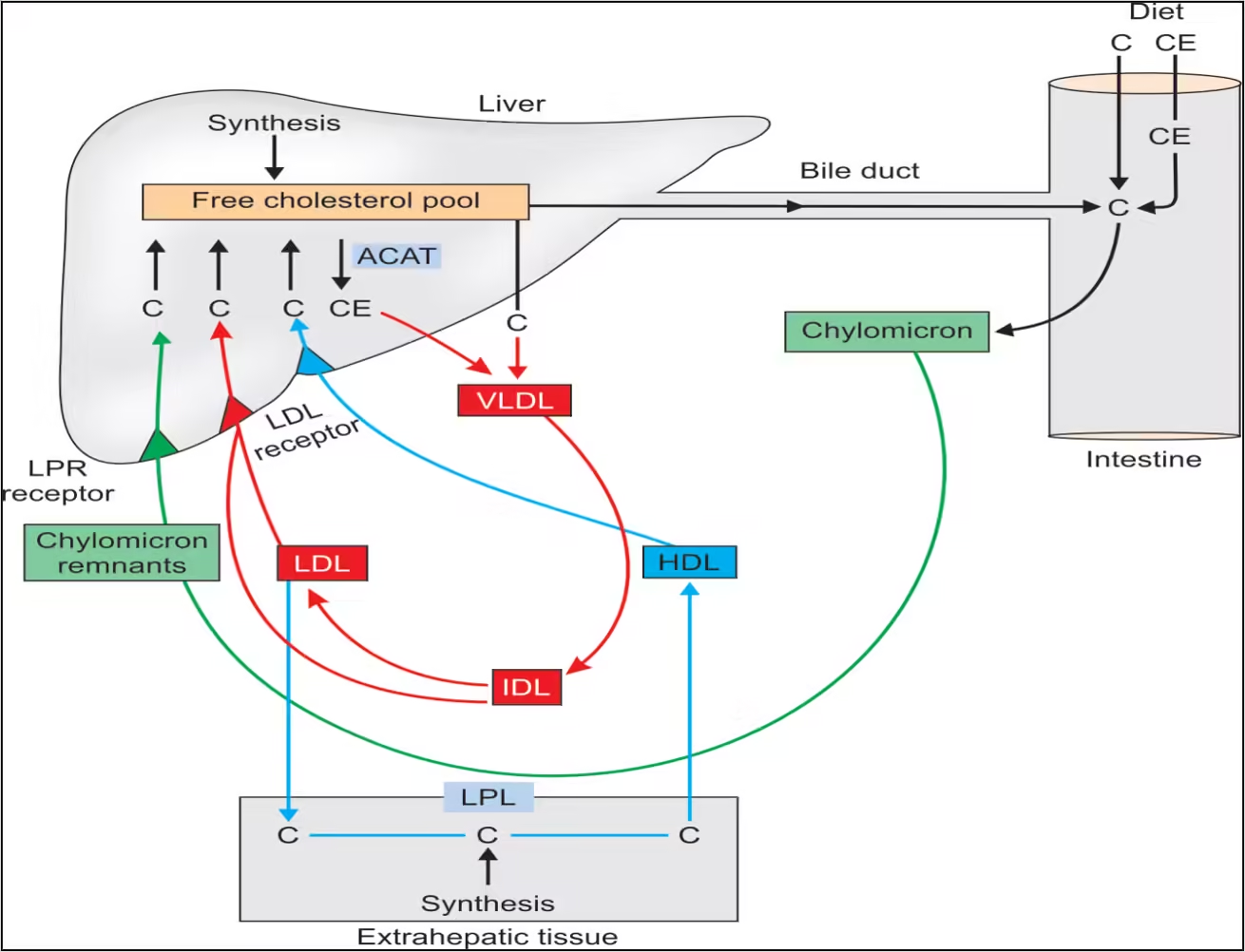
A. Lipoprotein Composition
- Low-Density Lipoprotein (LDL):
- Composed of a high percentage of cholesterol and triglycerides.
- It delivers cholesterol to peripheral tissues, and cells take it up via LDL receptors.
- High-Density Lipoprotein (HDL):
- It contains more protein than lipids and is involved in reverse cholesterol transport.
- Removes excess cholesterol from tissues and returns it to the liver.
- Very Low-Density Lipoprotein (VLDL):
- Rich in triglycerides, it transports lipids from the liver to tissues and is converted to LDL in circulation.
B. Receptor-Mediated Endocytosis
- LDL Receptor:
- Cells express LDL receptors that bind to apolipoprotein B-100 and E on LDL particles.
- Endocytosis occurs, leading to the uptake of LDL and the release of cholesterol into the cell for use or storage.
C. Reverse Cholesterol Transport
- Role of HDL:
- HDL particles scavenge excess cholesterol from peripheral tissues and transport it back to the liver.
- In the liver, cholesterol can be converted into bile acids or excreted into bile.
Regulation of Cholesterol Metabolism
Cholesterol homeostasis is maintained through complex regulatory mechanisms: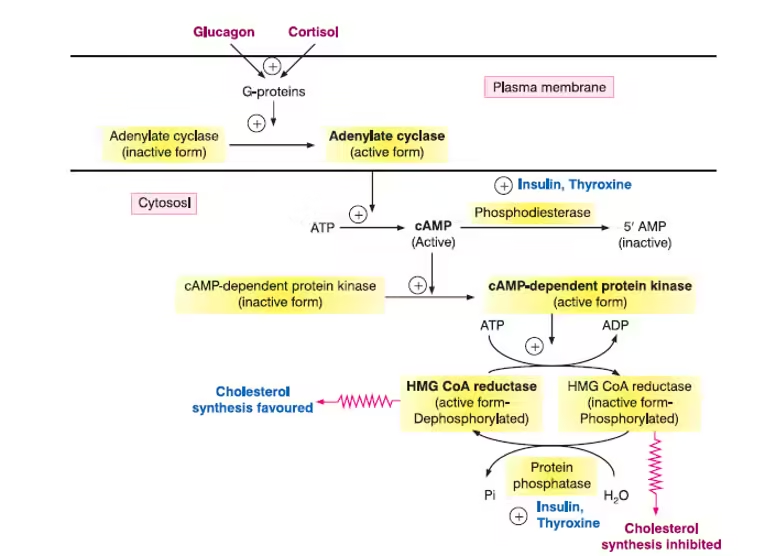
A. Feedback Inhibition
- SREBP Pathway:
-
- When cellular cholesterol levels are low, Sterol Regulatory Element-Binding Proteins (SREBPs) are activated, promoting gene expression in cholesterol synthesis and uptake (e.g., HMG-CoA reductase, LDL receptors).
- Insulin and Glucagon:
-
- Insulin stimulates cholesterol synthesis and uptake by promoting the activity of HMG-CoA reductase, while glucagon inhibits it.
B. Hormonal Influences
- Thyroid Hormones:
-
- Thyroid hormones can enhance cholesterol synthesis and LDL receptor activity, thus increasing cholesterol uptake.
- Statins and Other Drugs:
-
- Statins inhibit HMG-CoA reductase, reducing cholesterol synthesis and leading to upregulation of LDL receptors, which lowers circulating LDL levels.
Disorders of Cholesterol Metabolism
Disruptions in cholesterol metabolism can lead to various disorders, many of which have genetic underpinnings:
A. Familial Hypercholesterolemia (FH)
- Genetic Cause: Defects in the LDL receptor gene lead to impaired clearance of LDL cholesterol from the bloodstream.
- Symptoms:
- Extremely high LDL cholesterol levels (often >190 mg/dL).
- Early onset of atherosclerosis leads to cardiovascular diseases.
- Physical manifestations include xanthomas (cholesterol deposits in tendons and skin) and corneal arcus.
B. Atherosclerosis
- Mechanism: High levels of LDL cholesterol can accumulate in arterial walls, leading to plaque formation and vascular inflammation.
- Risk Factors: Genetics, diet, obesity, and lifestyle factors contribute to the development of atherosclerosis.
C. Cholesterol Gallstones
- Cause: Imbalances in bile components lead to precipitation of cholesterol, forming gallstones.
- Symptoms: Abdominal pain, nausea, vomiting, and complications if stones obstruct the bile duct.
D. Sitosterolemia
- Genetic Cause: Mutations in ABCG5 or ABCG8 genes increase the absorption of plant sterols (sitosterol) and cholesterol from the diet.
- Symptoms: Elevated cholesterol levels, premature atherosclerosis, and xanthomas.
E. Cholestasis
- Mechanism: Impaired bile flow due to liver disease or bile duct obstruction leads to the accumulation of bile acids and cholesterol in the liver.
- Symptoms: Jaundice, itching (pruritus), and fat malabsorption.
F. Niemann-Pick Disease Type C
- Genetic Cause: Mutations in genes responsible for lipid transport (e.g., NPC1).
- Symptoms: Accumulation of cholesterol and other lipids in cells, leading to neurological decline, hepatosplenomegaly, and early mortality.
Clinical Implications
- Diagnosis: Blood tests measure lipid profiles, identifying levels of total cholesterol, LDL, HDL, and triglycerides. Genetic testing can confirm specific disorders.
- Management Strategies:
- Lifestyle Changes: Dietary modifications (low saturated fat, high fiber), regular exercise, and weight management.
- Pharmacotherapy:
- Statins: Inhibit HMG-CoA reductase, reducing cholesterol synthesis and increasing LDL receptor expression.
- Bile Acid Sequestrants: Bind bile acids in the intestine, promoting bile acid excretion and lowering cholesterol levels.
- PCSK9 Inhibitors: Monoclonal antibodies inhibit PCSK9, increasing LDL receptor recycling and lowering LDL cholesterol.
HDL Metabolism
- High-density lipoprotein (HDL) is often called “good cholesterol” due to its role in reverse cholesterol transport, which helps remove excess cholesterol from tissues and prevent atherosclerosis.
- Here’s a comprehensive breakdown of HDL metabolism, including its formation, function, and regulatory mechanisms.
Biosynthesis of HDL
HDL is synthesized in two primary locations: the liver and the intestines.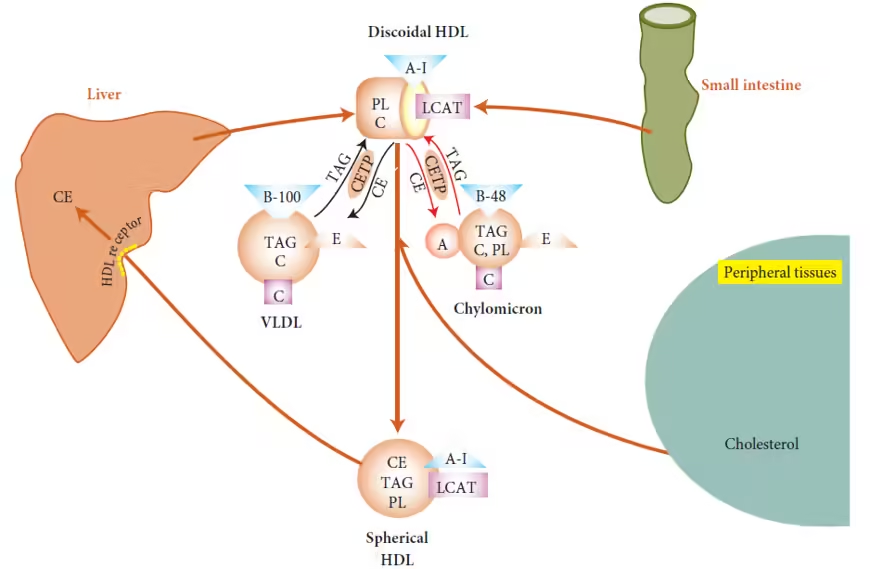
Liver:
- Nascent HDL Formation:
- HDL begins as lipid-poor particles composed mainly of proteins, particularly Apolipoprotein A-I (ApoA-I).
- The liver secretes these nascent HDL particles into the bloodstream.
Intestine:
- Chylomicron Remnants:
- After dietary fat absorption, the intestine forms chylomicrons, which transport triglycerides.
- When chylomicrons are hydrolyzed by lipoprotein lipase (LPL), remnants rich in cholesterol can transfer cholesterol to nascent HDL.
Maturation of HDL
Cholesterol Acquisition:
- Reverse Cholesterol Transport:
- HDL particles acquire free cholesterol from peripheral tissues through:
- ABCA1: This transporter facilitates cholesterol efflux from macrophages and other cells to nascent HDL.
- SR-BI (Scavenger Receptor Class B Type I): Mediates liver cells’ selective uptake of cholesteryl esters from HDL.
- HDL particles acquire free cholesterol from peripheral tissues through:
LCAT Activity:
- Lecithin-Cholesterol Acyltransferase (LCAT): This enzyme esterifies free cholesterol on HDL, converting it into more hydrophobic cholesteryl esters.
- As cholesteryl esters accumulate, they cause HDL to become larger and more lipid-rich, transitioning into mature HDL.
Function of HDL
Reverse Cholesterol Transport:
- HDL is crucial in removing excess cholesterol from peripheral tissues and transporting it back to the liver.
- This process helps prevent cholesterol accumulation in arterial walls, reducing the risk of atherosclerosis.
Antioxidant Properties:
- HDL has anti-inflammatory and antioxidant properties, helping to reduce oxidative stress and inflammation in the vascular system.
Endothelial Function:
- HDL promotes endothelial health by enhancing nitric oxide production, improving vascular tone, and inhibiting platelet aggregation.
HDL Clearance and Metabolism
Hepatic Uptake:
- The liver takes up HDL cholesterol via:
- SR-BI: This receptor allows for the selective uptake of HDL cholesteryl esters.
- After uptake, cholesterol can be repurposed for bile acid synthesis or excretion.
Lipid Transfer Proteins:
- CETP (Cholesterol Ester Transfer Protein): Facilitates the transfer of cholesteryl esters from HDL to other lipoproteins (like LDL and VLDL) in exchange for triglycerides.
- This process can affect HDL levels and its functionality in reverse cholesterol transport.
Regulation of HDL Metabolism
Dietary Influences:
- Healthy Fats: Monounsaturated and polyunsaturated fats (in olive oil, nuts, and fish) can increase HDL levels.
- Trans Fats: Consumption of trans fats can lower HDL levels.
Exercise:
- Regular physical activity is associated with increased HDL levels and improved HDL functionality.
Genetic Factors:
- Genetic variations can influence HDL levels, receptor function, and metabolism, contributing to individual variations in cardiovascular risk.
Pharmacological Interventions:
- Certain medications (e.g., niacin, fibrates) aim to raise HDL levels, although the impact on cardiovascular outcomes is still under investigation.
Clinical Implications of HDL Metabolism
- Dyslipidemia: Low HDL levels are a risk factor for cardiovascular diseases. Understanding HDL metabolism is crucial for developing strategies to manage lipid disorders.
- Cardiovascular Risk Assessment: HDL levels are often measured as part of lipid profiles to assess cardiovascular risk. Higher levels of HDL are generally associated with a lower risk of heart disease.
- Therapeutic Targets: Ongoing research aims to develop therapies that enhance HDL functionality, rather than merely increasing HDL levels, to improve cardiovascular outcomes.
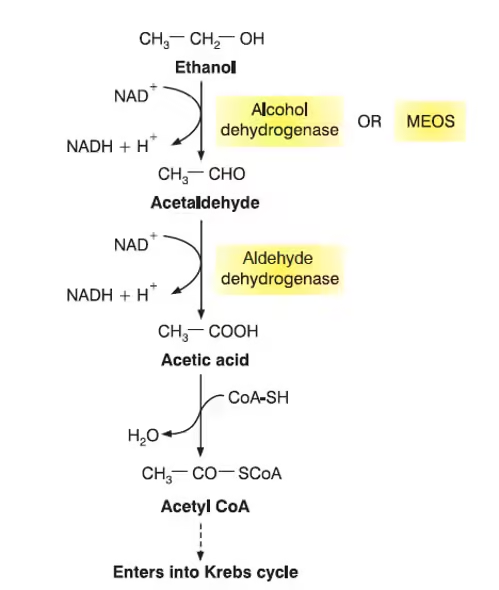
Alcohol metabolism
-
Alcohol metabolism refers to the biochemical processes by which the body breaks down and eliminates ethanol (ethyl alcohol) after its consumption.
-
The liver is the primary organ responsible for alcohol metabolism, where most ethanol is converted into less toxic substances.
-
The metabolism of alcohol occurs mainly through enzymatic oxidation reactions that convert ethanol into acetaldehyde and then into acetate.
-
The first step of alcohol metabolism involves the enzyme alcohol dehydrogenase (ADH), which converts ethanol into acetaldehyde, a toxic intermediate.
-
The second step involves aldehyde dehydrogenase (ALDH), which converts acetaldehyde into acetate, a less harmful compound.
-
Acetate is further converted into acetyl-CoA, which can enter metabolic pathways such as the citric acid cycle for energy production.
-
A small amount of alcohol is also metabolized by the microsomal ethanol oxidizing system (MEOS) and catalase pathway.
-
Excessive alcohol consumption can disturb normal metabolism and may lead to fatty liver, liver cirrhosis, and other metabolic disorders.