Introduction
- Phenylketonuria (PKU) is an inborn error of metabolism caused by a deficiency of the enzyme phenylalanine hydroxylase (PAH), which converts the amino acid phenylalanine to tyrosine.
- This leads to accumulation of phenylalanine and its toxic metabolites in blood and tissues.
- The disorder was first described by Asbjørn Følling in 1934, making it one of the earliest recognized metabolic diseases.
- PKU follows an autosomal recessive inheritance pattern and, if untreated, leads to severe intellectual disability, microcephaly, epilepsy, and other neurodevelopmental abnormalities.
- Early detection through newborn screening and dietary management prevents these complications almost entirely.
Signs and Symptoms
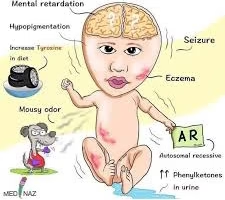
Phenylketonuria manifests when elevated levels of phenylalanine and its metabolites accumulate in the body, exerting toxic effects on the developing nervous system. The clinical features vary based on the degree of enzyme deficiency, dietary control, and age of onset.
1. Neurological Manifestations
-
Intellectual disability: Progressive and irreversible if untreated; typically becomes evident after 4–6 months of age.
-
Developmental delay: Delayed milestones, poor motor coordination, and slow learning.
-
Seizures: Often generalized tonic–clonic or myoclonic in nature.
-
Behavioral abnormalities: Hyperactivity, irritability, restlessness, and self-injurious behavior.
-
Psychiatric symptoms: Depression, anxiety, attention deficit, and executive function deficits may occur even with treatment lapses.
-
Tremors and spasticity: Due to impaired neurotransmitter synthesis and abnormal myelination.
2. Physical Features
-
Microcephaly: Reduced head circumference due to brain hypomyelination.
-
Fair skin, light hair, and blue eyes: Owing to decreased tyrosine availability for melanin synthesis.
-
Eczema and skin rashes: Common dermatological findings resembling atopic dermatitis.
-
Failure to thrive: Poor weight gain and stunted growth if untreated.
3. Sensory and Cognitive Impairments
-
Speech delay and language disorder: Often one of the earliest signs.
-
Reduced IQ and cognitive performance: Correlates directly with duration of elevated phenylalanine exposure.
-
Impaired executive functions: Difficulties in planning, working memory, and attention.
4. Characteristic Odor
-
The presence of “mousy” or musty body odor is a hallmark sign, resulting from excretion of phenylacetate and other aromatic metabolites in urine, sweat, and breath.
5. Behavioral and Psychological Features
-
Social withdrawal and poor emotional regulation may occur.
-
Anxiety and mood disturbances often persist even in treated adults.
-
Autism-like traits have been described in severe untreated cases.
6. In Treated Individuals
-
Even with strict dietary control, some may exhibit mild cognitive, attention, and mood disorders, likely due to chronic fluctuations in plasma phenylalanine or genetic modifiers of PAH activity.
Genetics
1. Gene and Chromosomal Location
-
The PAH gene is located on chromosome 12q23.2, spanning approximately 90 kilobases and consisting of 13 exons.
-
It encodes the phenylalanine hydroxylase enzyme, which catalyzes the hydroxylation of phenylalanine to tyrosine—a critical step in amino acid metabolism.
-
The enzyme functions primarily in the liver and to a lesser extent in the kidney.
2. Inheritance Pattern
-
Autosomal recessive inheritance: Both alleles of the PAH gene must carry a pathogenic variant for an individual to manifest PKU.
-
Carriers (heterozygotes) are asymptomatic but have ~50% of normal enzyme activity.
-
When both parents are carriers:
-
25% chance of an affected child (homozygous mutant),
-
50% chance of a carrier child,
-
25% chance of a normal child.
-
3. Mutations and Molecular Defects
-
Over 1000 distinct mutations in the PAH gene have been identified.
-
The most common mutation types include:
-
Missense mutations – cause single amino acid substitutions affecting enzyme folding or activity.
-
Nonsense mutations – introduce premature stop codons leading to truncated, inactive enzymes.
-
Splice-site mutations – disrupt mRNA processing.
-
Deletions or insertions – result in frame shifts and loss of function.
-
-
Example: The R408W mutation (arginine → tryptophan at codon 408) is one of the most frequent worldwide, especially in European populations.
4. Genotype–Phenotype Correlation
-
The severity of PKU depends on residual enzymatic activity of PAH:
-
<1% activity: Classic PKU – severe hyperphenylalaninemia (>1200 µmol/L).
-
1–5% activity: Moderate or mild PKU.
-
>5% activity: Mild hyperphenylalaninemia (non-PKU type).
-
-
Compound heterozygotes (different mutations on each allele) often display intermediate phenotypes.
-
This variability explains why some patients respond to BH₄ (sapropterin) therapy — those with partially active enzymes may regain function when BH₄ availability is enhanced.
5. Other Genetic Causes of Hyperphenylalaninemia
Although PAH gene mutations cause classical PKU, 2–3% of cases result from defects in tetrahydrobiopterin (BH₄) metabolism, leading to BH₄-deficient hyperphenylalaninemia.
-
Genes involved:
-
GCH1 – GTP cyclohydrolase I
-
PTS – 6-pyruvoyl tetrahydropterin synthase
-
QDPR – dihydropteridine reductase
-
PCBD1 – pterin-4α-carbinolamine dehydratase
-
-
These variants lead to combined deficiencies of dopamine and serotonin synthesis due to impaired cofactor regeneration.
6. Molecular Diagnosis
-
DNA-based testing (e.g., Sanger sequencing or next-generation sequencing) identifies PAH mutations to confirm diagnosis, assess carrier status, and predict therapeutic response.
-
Prenatal diagnosis can be performed using chorionic villus sampling or amniocentesis if both parental mutations are known.
-
Carrier screening is now incorporated into many population-based genetic panels due to the disease’s preventability.
7. Genetic Counselling
-
Essential for families with a history of PKU.
-
Parents are counseled on the 25% recurrence risk and the importance of newborn screening for early intervention.
-
Maternal PKU counseling is vital since high maternal phenylalanine levels are teratogenic even if the fetus is heterozygous.
Pathophysiology
1. Normal Phenylalanine Metabolism
Under physiological conditions, dietary phenylalanine, an essential amino acid, is metabolized via the following pathway:
Phenylalanine → Tyrosine → DOPA → Dopamine → Norepinephrine → Epinephrine
-
-
The conversion of phenylalanine to tyrosine is catalyzed by the hepatic enzyme phenylalanine hydroxylase (PAH).
-
This reaction requires three key cofactors:
-
Tetrahydrobiopterin (BH₄) – a reducing cofactor.
-
Molecular oxygen (O₂) – donates oxygen atoms.
-
Iron (Fe²⁺) – at the enzyme’s active site for catalysis.
-
-
The reaction produces tyrosine, which serves as a precursor for catecholamines, thyroid hormones, and melanin.
-
2. Primary Biochemical Defect
In PKU, mutations in the PAH gene cause partial or complete loss of enzyme activity, leading to:
-
-
Accumulation of phenylalanine in blood and tissues.
-
Decreased synthesis of tyrosine and its downstream metabolites.
-
When phenylalanine levels rise above the normal plasma range (>120 µmol/L; normal <120 µmol/L), alternative metabolic pathways become active. The excess phenylalanine is transaminated to form toxic metabolites such as:
-
-
Phenylpyruvate
-
Phenylacetate
-
Phenyllactate
-
These organic acids are collectively known as phenylketones, and their urinary excretion gives the condition its name: phenylketonuria.
3. Secondary Neurochemical Consequences
Elevated phenylalanine has multiple neurotoxic and metabolic effects:
a. Disruption of Brain Amino Acid Transport
-
-
Phenylalanine competes with other large neutral amino acids (LNAAs) — tyrosine, tryptophan, leucine, isoleucine, and valine — for transport across the blood–brain barrier (BBB).
-
High plasma phenylalanine saturates the LAT1 transporter, reducing cerebral uptake of LNAAs needed for protein and neurotransmitter synthesis.
-
b. Neurotransmitter Deficiency
-
-
Tyrosine deficiency → ↓ dopamine and norepinephrine (catecholamines).
-
Tryptophan deficiency → ↓ serotonin.
-
Result: Impaired myelination, delayed brain development, and abnormal neuronal signaling leading to cognitive impairment and mood disturbances.
-
c. Impaired Myelination and White Matter Damage
-
-
High phenylalanine interferes with oligodendrocyte function and lipid synthesis, causing hypomyelination of the brain.
-
MRI studies reveal white matter hyperintensities, particularly in the parietal and occipital lobes.
-
d. Oxidative Stress
-
-
Excess phenylalanine generates reactive oxygen species (ROS) and impairs antioxidant defense, causing neuronal oxidative damage.
-
Reduced glutathione and catalase activities have been documented in untreated PKU patients.
-
4. Role of Tetrahydrobiopterin (BH₄)
-
BH₄ acts as a cofactor for PAH, tyrosine hydroxylase, and tryptophan hydroxylase.
-
In BH₄-deficient hyperphenylalaninemia, phenylalanine accumulates due to defective BH₄ synthesis or regeneration — not because of a PAH mutation.
-
This also causes deficiency of dopamine and serotonin, resulting in more severe neurological manifestations such as hypotonia, movement disorders, and autonomic dysfunction.
5. Tyrosine Deficiency and Hypopigmentation
-
Since tyrosine is a precursor for melanin, reduced conversion from phenylalanine causes:
-
Fair skin and light-colored hair.
-
Blue irides due to lack of melanin deposition.
-
Eczema-like dermatitis caused by impaired skin barrier and nutrient deficiency.
-
6. Systemic Manifestations
-
Metabolic acidosis: due to organic acid accumulation (phenylpyruvate, phenyllactate).
-
“Mousy odor”: from phenylacetate excretion in urine, sweat, and breath.
-
Endocrine and growth disturbances: from protein restriction and amino acid imbalance if diet is poorly managed.
7. Neuropathological Changes
Histopathological studies of untreated PKU brains show:
-
Hypomyelination and cortical atrophy.
-
Gliosis and neuronal loss, especially in the white matter and basal ganglia.
-
Reduced dendritic arborization and synaptic density, explaining cognitive deficits.
Metabolic Pathways
1. Normal Phenylalanine Metabolic Pathway
-
Enzyme:
-
The reaction is catalyzed by phenylalanine hydroxylase (PAH), located mainly in the liver.
-
-
Cofactors Required:
-
Tetrahydrobiopterin (BH₄) acts as an electron donor.
-
Oxygen (O₂) donates one oxygen atom to the hydroxyl group of phenylalanine and one to water.
-
Iron (Fe²⁺) at the active site assists in redox reactions.
-
-
Products Formed:
-
Tyrosine, which becomes a precursor for catecholamines (dopamine, norepinephrine, epinephrine), thyroid hormones, and melanin.
-
Dihydrobiopterin (BH₂), which is regenerated back to BH₄ by dihydropteridine reductase (DHPR) using NADH.
-
2. Alternate Pathways in PKU (Defective PAH)
When PAH enzyme activity is deficient, phenylalanine accumulates and diverts to secondary (minor) metabolic routes:
a. Transamination Pathway:
Phenylalanine + α-ketoglutarate → Phenylpyruvate + Glutamate
(Catalyzed by aminotransferase enzyme)
b. Reduction and Decarboxylation Pathways:
-
-
Phenylpyruvate → Phenyllactate (via lactate dehydrogenase)
-
Phenylpyruvate → Phenylacetate (via decarboxylation)
-
c. Excretion:
These metabolites — phenylpyruvate, phenyllactate, and phenylacetate — are collectively called “phenylketones.” They accumulate in blood and are excreted in urine and sweat, producing the characteristic “mousy odor”.
3. Downstream Effects of Blocked Pathway
-
Reduced Tyrosine:
Tyrosine becomes a conditionally essential amino acid in PKU.
↓ Tyrosine → ↓ DOPA → ↓ Dopamine → ↓ Norepinephrine → ↓ Epinephrine. -
Neurotransmitter Deficiency:
Leads to impaired synaptic signaling and neurological dysfunction. -
Hypopigmentation:
Tyrosine deficiency reduces melanin synthesis, leading to light hair and skin. -
Neurotoxicity:
Excess phenylalanine interferes with transport of large neutral amino acids (LNAAs) across the blood–brain barrier, disturbing brain development.
4. BH₄-Dependent Pathways (Cofactor Role)
BH₄ is required for three hydroxylase enzymes:
-
Phenylalanine hydroxylase (PAH) → converts phenylalanine to tyrosine.
-
Tyrosine hydroxylase (TH) → converts tyrosine to DOPA.
-
Tryptophan hydroxylase (TPH) → converts tryptophan to 5-hydroxytryptophan.
Diagnosis
1. Newborn Screening
-
Purpose: Early detection before clinical symptoms appear.
-
Timing: Blood sample collected 24–72 hours after birth, ideally after the infant has begun feeding (since phenylalanine levels rise with protein intake).
-
Specimen: A few drops of heel-prick blood blotted on filter paper (Guthrie card).
Screening Methods:
-
Guthrie Bacterial Inhibition Assay (Classical Method):
-
Introduced in 1963 by Robert Guthrie.
-
Based on the ability of elevated phenylalanine in the blood to overcome inhibition of Bacillus subtilis growth caused by β-2-thienylalanine.
-
A positive test shows bacterial growth around the sample spot.
-
-
Tandem Mass Spectrometry (MS/MS) – Modern Method:
-
Quantitatively measures phenylalanine and tyrosine concentrations in dried blood spots.
-
More sensitive and can detect multiple metabolic disorders simultaneously.
-
Phenylalanine/Tyrosine ratio >3 is suggestive of PKU.
-
2. Confirmatory Testing
If the screening test is positive, confirmatory diagnostic steps are undertaken:
| Test | Principle / Purpose | Diagnostic Finding |
|---|---|---|
| Plasma Amino Acid Analysis (HPLC / MS/MS) | Quantitative measurement of phenylalanine and tyrosine | Phenylalanine >120 µmol/L (normal <120 µmol/L); Classic PKU often >1200 µmol/L |
| Urine Organic Acid Analysis (GC-MS) | Detects abnormal metabolites | Presence of phenylpyruvate, phenylacetate, phenyllactate |
| BH₄ Loading Test | Differentiates classical PAH deficiency from BH₄ deficiency | >30% reduction in Phe level after BH₄ administration → BH₄-responsive |
| PAH Gene Analysis | Identifies specific mutations | Confirms diagnosis and guides treatment (e.g., BH₄ responsiveness) |
| Enzyme Assay (Liver Biopsy, rarely done) | Measures phenylalanine hydroxylase activity | Decreased or absent enzyme activity |
3. Differential Diagnosis
Other causes of hyperphenylalaninemia (HPA) should be ruled out:
-
Tetrahydrobiopterin (BH₄) deficiency
-
Dihydropteridine reductase deficiency
-
Liver disease
-
Prematurity or transient neonatal HPA
These conditions are identified by normal PAH gene with abnormal pterin profile or enzyme cofactor assays.
4. Diagnostic Criteria
| Parameter | Normal | Mild HPA | Mild PKU | Classical PKU |
|---|---|---|---|---|
| Plasma Phenylalanine (µmol/L) | <120 | 120–600 | 600–1200 | >1200 |
| Tyrosine Level | Normal | Normal or low | Low | Markedly low |
| Urinary Phenylketones | Absent | Trace | Present | Markedly increased |
| Clinical Features | None | None / Mild | Mild neurocognitive signs | Severe neurological impairment if untreated |
5. Prenatal Diagnosis
-
Indicated in families with known PAH mutations.
-
Methods:
-
Chorionic villus sampling (CVS) at 10–12 weeks or amniocentesis at 15–18 weeks.
-
DNA extracted from fetal cells analyzed for PAH gene mutations.
-
Non-invasive prenatal testing (NIPT) using maternal blood cell-free DNA is under study.
-
6. Neuroimaging and Supportive Investigations
-
MRI Brain: In untreated PKU, shows diffuse white matter changes, hypomyelination, and parietal lobe hyperintensities.
-
EEG: May reveal generalized spike–wave discharges in cases with seizures.
-
Neuropsychological Testing: For evaluating cognitive outcomes during follow-up.
7. Importance of Early Detection
-
Early identification and prompt dietary management (within the first 10–14 days of life) prevent almost all neurological manifestations.
-
Delayed diagnosis beyond the first month of life often results in irreversible intellectual disability.
-
Continuous lifelong monitoring of blood phenylalanine levels ensures optimal neurocognitive development.
8. Follow-up Monitoring
-
Frequency:
-
Infants: Weekly to biweekly.
-
Children: Monthly.
-
Adults: Every 3–6 months.
-
-
Targets: Maintain plasma phenylalanine between 120–360 µmol/L (children) and <600 µmol/L (adults).
-
Methods: Finger-prick dried blood spot testing or capillary plasma testing.
Treatment
1. Principles of Treatment
-
Lower plasma phenylalanine levels to prevent neurotoxicity.
-
Maintain adequate tyrosine levels (since it becomes conditionally essential).
-
Support normal growth, neurodevelopment, and cognitive function.
-
Avoid overt restriction that can lead to protein malnutrition.
-
Ensure lifelong management, as even adults are vulnerable to neuropsychological effects if levels rise.
2. Dietary Therapy
Dietary restriction of phenylalanine is the cornerstone of PKU management.
a. Low-Phenylalanine Diet
-
-
Objective: Keep plasma Phe between 120–360 µmol/L in children and <600 µmol/L in adults.
-
Restricted foods: Meat, fish, eggs, milk, cheese, nuts, soy, pulses, and artificial sweeteners containing aspartame.
-
Allowed foods: Fruits, vegetables, sugars, fats, and special low-protein products.
-
Medical foods: Special Phe-free amino acid formulas provide necessary nutrients without phenylalanine.
-
b. Timing of Initiation
-
-
Must begin within the first 7–10 days of life for best neurodevelopmental outcomes.
-
Delay beyond 3 weeks increases risk of irreversible brain injury.
-
c. Special Low-Protein Foods
Bread, pasta, rice, biscuits, and flour substitutes specifically designed for PKU patients to maintain calorie intake.
3. Nutritional Supplements
a. Tyrosine
-
-
Supplemented as it becomes conditionally essential in PKU.
-
Tyrosine is vital for the synthesis of catecholamines, thyroid hormones, and melanin.
-
b. Vitamins and Minerals
-
-
Deficiencies may occur due to dietary restrictions; supplementation of the following is important:
-
Vitamin B12, folate, iron, zinc, selenium, and calcium.
-
Omega-3 fatty acids (DHA and EPA) for brain and visual development.
-
-
c. Large Neutral Amino Acids (LNAA)
-
-
Supplements containing tyrosine, tryptophan, valine, leucine, and isoleucine compete with phenylalanine at the blood–brain barrier, reducing its uptake into the brain.
-
Beneficial for adolescents and adults with suboptimal dietary adherence.
-
4. Pharmacologic and Enzyme-Based Therapies
a. Sapropterin Dihydrochloride (BH₄ Analog)
-
-
Mechanism: Synthetic form of tetrahydrobiopterin (BH₄), the natural cofactor for phenylalanine hydroxylase (PAH).
-
Use: Effective in patients with residual PAH activity (BH₄-responsive PKU).
-
Effect: Lowers blood phenylalanine by enhancing enzyme activity.
-
Dose: 10–20 mg/kg/day orally.
-
Response: Monitored by measuring Phe levels after 24–48 hours.
-
Advantages: Expands diet tolerance and improves quality of life.
-
b. Pegvaliase (Palynziq®)
-
-
Type: Enzyme substitution therapy using PEGylated phenylalanine ammonia lyase (PAL).
-
Mechanism: Converts phenylalanine → trans-cinnamic acid + ammonia, bypassing PAH.
-
Indication: Adults (>18 years) with uncontrolled phenylalanine levels (>600 µmol/L) despite diet and BH₄ therapy.
-
Route: Subcutaneous injection.
-
Caution: Risk of anaphylaxis, requires gradual dose escalation under medical supervision.
-
c. Gene Therapy (Experimental)
-
-
Trials using adeno-associated viral (AAV) vectors to deliver functional PAH gene to liver cells have shown promising results in animals, but are still under research.
-
Treatment in Mothers (Maternal PKU)
Women with PKU must maintain tight metabolic control before conception and throughout pregnancy to prevent maternal PKU syndrome in the fetus.
a. Risks to the Fetus:
-
-
Microcephaly
-
Congenital heart defects
-
Intrauterine growth retardation
-
Intellectual disability
-
Low birth weight
-
b. Management:
-
-
Strict low-Phe diet initiated before conception (ideally 3 months prior).
-
Frequent monitoring: Plasma phenylalanine every 1–2 weeks during pregnancy.
-
Maintain Phe levels between 120–360 µmol/L.
-
Nutritional support: Ensure adequate intake of tyrosine, folate, and DHA for fetal neurodevelopment.
-
Monitoring and Follow-Up
-
Infants: Weekly to biweekly plasma phenylalanine measurements.
-
Children: Monthly monitoring.
-
Adults: Every 3–6 months.
-
Tools: Dried blood spot testing or plasma amino acid profiling.
-
Clinical assessments: Neurocognitive testing, growth tracking, and dietary adherence evaluation.
| Parameter | Target Range | Age Group |
|---|---|---|
| Plasma Phe | 120–360 µmol/L | Infants/Children |
| Plasma Phe | <600 µmol/L | Adults |
| Tyrosine | 40–100 µmol/L | All ages |